Вычислительный конвейер MultiDeNA
Ссылка на приложение Galaxy
https://galaxy.icgbio.ru/root?tool_id=multidena
MultiDeNA
Программный комплекс MultiDeNa (Tsukanov et al., 2022) позволяет сочетать методологически разные модели de novo поиска мотивов, а именно традиционную модель PWM, не учитывающую зависимости позиций мотива, и также альтернативные модели (InMoDe (Eggeling et al., 2017), BaMM (Ge et al., 2021), SiteGA (Tsukanov et al., 2022), предлагающие разные подходы для выявления зависимостей нуклеотидного контекста мотива. «MultiDeNA» является конвейером для унифицированной обработки данных ChIP-seq, включая применение традиционной PWM и альтернативных (BaMM, SiteGA и др.) моделей мотивов. В конвейере MultiDeNa реализованы:
(1) оценка точности моделей с помощью перекрёстной проверки (two fold cross-validation), которая позволяет произвести подбор оптимальных параметров моделей (например, длина мотива, порядок марковской модели и др. в зависимости от модели);
(2) процедура для единообразной установки порогов распознавания сайтов связывания для каждой модели;
(3) процедура поиска сайтов связывания каждой моделью;
(4) процедура объединения результатов распознавания всех моделей.
Дополнительно MultiDeNA позволяет провести анализ терминов генной онтологии на основании результатов распознавания, а также сравнить полученные de novo мотивы с известными с помощью TomTom. Общая схема работы конвейера показана на схеме ниже (рис. 1).
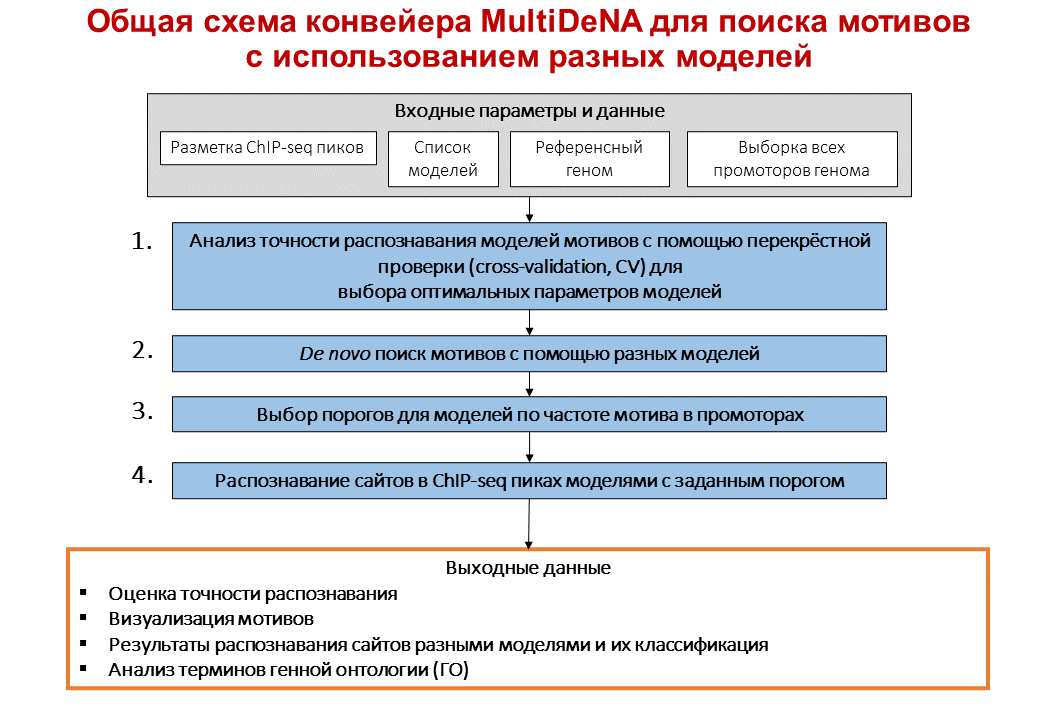
Рис. 1. Описание основных этапов работы конвейера MultiDeNA
Программный модуль «MultiDeNa» позволяет выявлять список предсказанных сайтов связывания (СС) транскрипционных факторов (ТФ), за счёт применения нескольких методологически различных моделей мотивов. Программный модуль «MultiDeNa» можно использовать в исследованиях по анализу ChIP-seq экспериментов, с его помощью можно расширить список генов мишеней ТФ, и тем самым прояснить механизмы регуляции транскрипции генов с помощью ТФ. Параметры, которые задаются для запуска MultiDeNA приведены ниже на рисунке 2.
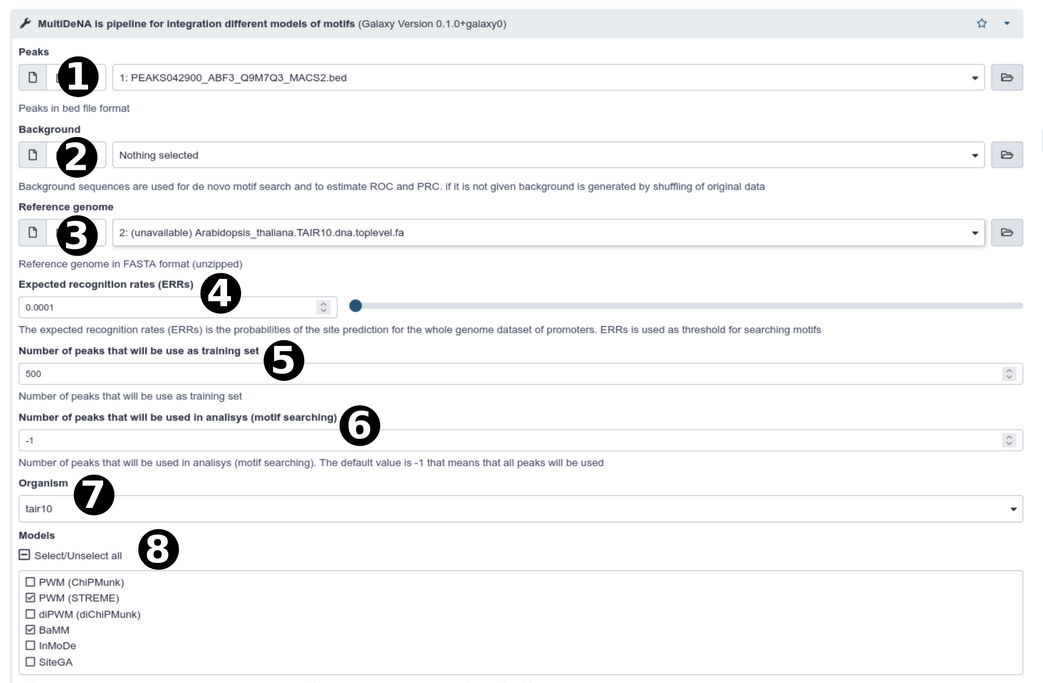
Рис. 2. Пример запуска MultiDeNA с помощью Galaxy. Основные параметры: 1 (Peaks) - пики ChIP-seq в виде bed разметки (позитивная выборка); 2 (Background) - негативная выборка в fasta формате; 3 (Reference genome) - референсный геном в fasta формате; 4 (Expected Recognition Rate) - частота встречаемости мотива (ERR) в промоторах всех генов, кодирующих белки, эта величина позволяет рассчитать порог распознавания модели; 5 - Количество пиков на которых будет проводится обучение моделей (de novo поиск) и оценка точности (построение ROC-кривой); 6 - Количество пиков в которых будут искаться сайты связывания уже обученными моделями, если параметр равен -1, то используются все пики, что есть в данных; 7 (Organism) – Вид организма, для которого были получены пики ChIP-seq; 8 (Models) – Модели мотива, которые будут использоваться в анализе. Параметры от 1 до 5 и 7, 8 являются общими для перекрёстной проверки и обучения модели. Параметр 6 отражает применение модели, полученной после перекрёстной проверки и последующем обучении.
Основные этапы работы «MultiDeNa» описаны в презентации.
Комментарии для запуска MultiDeNA
- Для запуска «MultiDeNa» в качестве основных входных данных используется пики ChIP-seq в виде bed разметки и референсный геном в fasta формате, того вида организма, для которого были получены пики. Чтобы загрузить данные нажмите на “Upload Data”
-
После того как данные загружены на сервер, нужно выбрать эти файлы в качестве параметров для «MultiDeNa». В окне 1 (Peaks, см. схему) выберете файл в bed формате. Далее в окне 3 (Reference genome, см. схему) необходимо выбрать выпуск генома в формате fasta.
-
На этапе оценки точности и этапе de novo поиска используется негативная выборка (background), которая играет роль контраста для позитивной выборки при de novo поиске. Негативную выборку можно получить путём перемешивания нуклеотидов последовательностей позитивной выборки или из полного генома с помощью модуля «AntiNoise». Если вы получили и загрузили негативную выборку, то добавьте её в окне 2 (Background, см. схему).
-
Выбор порога распознавания сайтов по величине частоты встречаемости мотива (Expected recognition rate), который задается в окне 4 (Expected recognition rate, см. схему, рис. 2). По умолчанию для всех моделей порог выставляется по частоте мотива (ERR) в промоторах равный 0.0001, это “средний” порог распознавания. Вы можете менять значение ERR, при увеличении до 0.0005 модели будут распознавать больше сайтов, но при этом увеличивается ошибка перепредсказания. Рекомендуем использовать ERR в диапазоне от 0.00001 до 0.001.
-
Выбор количества пиков для позитивной выборки (выборки обучения) в окне 5 (см. схему). Все пики что подаются в «MultiDeNa» будут отсортированы по качеству, от лучших к худшим. Первые N будут использоваться для оценки точности моделей, выбора оптимальных параметров моделей и de novo поиск мотивов на оптимальных параметрах каждой модели. По умолчанию N = 500, не рекомендуем уменьшать это число, для получения более корректных результатов лучше использовать N = 1000 или более, но увеличение этого числа будет приводить к увеличению времени расчёта.
-
Выбор количества пиков в которых будут искаться сайты связывания после того как модели были обучены (после de novo поиска) в окне 6 (см. схему). По умолчанию сайты распознаются во всех пиках
S = -1, т.е. это все пики включая те, что использовались и при обучении (предыдущий пункт). Если S = 1200 и N = 500, то в S входят 500 пиков, которые были в обучении и следующие 700 пиков. -
Выбор вида организма для которого был сделан ChIP-seq эксперимент в окне 7 (Organism). Этим параметром выбираются промоторы, на которых будет оцениваться частота встречаемости мотивов для каждой модели для всех порогов и рассчитываться таблица порогов, которая необходима, чтобы выбрать порог модели по заданному ERR из пункта 4.
-
Выбор моделей, которые будут использоваться в анализе в окне 8 (Models). По умолчанию выбраны две модели PWM (Streme) и BaMM. Для работы «MultiDeNa» необходимо, как минимум две модели. Модель BaMM работает только тогда, когда используется одна из моделей PWM, так как для её работы необходима модель PWM.
-
Дополнительно можно задать базу мотивов в формате meme. База мотивов будет использоваться для сравнения de novo мотивов с известными. Данный параметр опциональный. Базу мотивов в формате meme можно взять из HOCOMOCO и JASPAR.
Описание выходных данных
После того, как «MultiDeNa» закончит работу результаты можно будет скачать в виде архива. Ниже приведена структура выходного архива, в котором содержаться результаты работы «MultiDeNa» (в качестве моделей были использованы PWM (Streme) и BaMM):
├── annotation
│ ├── test
│ │ ├── all_models_GO.tsv
│ │ ├── bamm_annotaion.tsv
│ │ ├── compare_model_GO.tsv
│ │ ├── pwm_annotaion.tsv
│ │ ├── pwm_model_GO.tsv
│ │ └── tools_annotaion.pdf
│ └── train
│ ├── bamm_annotaion.tsv
│ ├── compare_models_GO.tsv
│ ├── pwm_annotaion.tsv
│ ├── pwm_model_GO.tsv
│ └── tools_annotaion.pdf
├── auc
│ ├── bamm
│ │ ├── statistics.txt
│ │ ├── ...
│ └── pwm
│ ├── statistics.txt
│ ├── ...
├── bed
├── fasta
├── models
│ ├── bamm_model
│ │ ├── bamm.hbcp
│ │ ├── bamm_model.ihbcp
│ │ ├── bootstrap_merged.txt
│ │ └── bootstrap.txt
│ ├── pwm_model
│ │ ├── bootstrap_merged.txt
│ │ ├── bootstrap.txt
│ │ ├── pwm_model.meme
│ │ ├── pwm_model.pfm
│ │ ├── pwm_model.pwm
│ │ ├── streme.txt
│ │ └── streme.xml
│ └── thresholds
│ ├── bamm_model_thresholds.txt
│ └── pwm_model_thresholds.txt
├── results
│ ├── combined_scan_test_1.90e-04.pro
│ ├── combined_scan_train_1.90e-04.pro
│ ├── compare_motif_logo.pdf
│ ├── compare_test_1.90e-04_pwm.bamm_counts.tsv
│ ├── compare_train_1.90e-04_pwm.bamm_counts.tsv
│ ├── peaks_classification_test_1.90e-04.tsv
│ └── peaks_classification_train_1.90e-04.tsv
├── scan
│ ├── bamm_test_1.90e-04.bed
│ ├── bamm_train_1.90e-04.bed
│ ├── pwm_test_1.90e-04.bed
│ └── pwm_train_1.90e-04.bed
├── scan-best
│ ├── bamm.scores.txt
│ └── pwm.scores.txt
└── tomtom
├── bamm.meme
├── bamm.pfm
├── bamm.pwm
├── bamm.sites.txt
├── bamm.tomtom_results
│ ├── tomtom.tsv
│ └── tomtom.xml
├── pwm.meme
├── pwm.pfm
├── pwm.pwm
├── pwm.sites.txt
└── pwm.tomtom_results
├── tomtom.tsv
└── tomtom.xml
Основные результаты «MultiDeNa» находятся в разделе results.
Раздел results
- combined_scan_{train/test}_{err} - разметка в формате pro (гибрид bed и fasta форматов) в которой записаны результаты распознавания ССТФ каждой модели.
Пример:
\>peaks\_0::chr1:164712189-164712712(+) SEQ 1 \#\# \<- first peak
162 170 4.137341427518557 + gataacgg bamm 1 \#\# \<- start end
-log10(err) strand site model\_name group (if several models have the
same group they are overlapped)
234 242 3.7438654156944446 + gataacag bamm 2
245 265 3.740915004460945 - cgagcgataaggctgctaac sitega 3
250 262 3.8460276036525065 - gcgataaggctg pwm 3
252 260 3.9555972318346377 - gataaggc bamm 3
357 377 4.715008011604894 - cttccgatatcaaccgggaa sitega 4
440 460 4.364457279299288 - atggtgattaacatccccct sitega 5
\>peaks\_1::chr3:93470273-93470886(+) SEQ 2 \#\# \<- second peak
106 126 3.9101503930828088 + actttgatatttcatgtaga sitega 1
\>peaks\_2::chr17:36862394-36862756(+) SEQ 3
129 137 3.7257217215553267 + gataacac bamm 1
193 205 3.8499146293487376 + cagataaccgag pwm 2
195 203 3.775843524673776 + gataaccg bamm 2
В начале записи идёт заголовок >peaks_0::chr1:164712189-164712712(+) SEQ 1 - это информация о пике в котором были распознаны сайты. Далее в формате bed перечислены распозанные сайты:
------------------ ----------------- -------------------- ------ ---------------------- -------- ------------
Начало сайта (1) Конец сайта (1) -log10(ERR) Цепь Сайт Модель Группа (2)
162 170 4.137341427518557 \+ gataacgg bamm 1
234 242 3.7438654156944446 \+ gataacag bamm 2
245 265 3.740915004460945 \- cgagcgataaggctgctaac sitega 3
250 262 3.8460276036525065 \- gcgataaggctg pwm 3
252 260 3.9555972318346377 \- gataaggc bamm 3
------------------ ----------------- -------------------- ------ ---------------------- -------- ------------
(1) - Координаты сайта даны относительно начала последовательности пика, а не всего генома
(2) - Номер группы пересекающихся сайтов, предсказанных разными моделями. Группы содержат сайты с перекрывающейся локализацией, предсказанные разными моделями. Наличие групп определяется разными оптимальными параметрами моделей, такими как длина мотива.
-
compare_motif_logo.pdf - файл содержит лого моделей
-
compare_{train/test}_{err}_{model1}.{model2}_counts.tsv - файл в котором записаны результаты пересечения {model1} и {model2}, при этом пересечение считается с учётом того, что сайты предсказанные разными моделями могут пересечься. Данные файлы будут получены для каждой пары моделей, если моделей больше двух.
Пример:
------------------- ------------------- --------------------- -------------------------- -------------------- -------------------
pwm: against bamm bamm: against pwm overlapped:pwm,bamm not\_overlapped:pwm,bamm no\_sites:pwm,bamm number\_of\_peaks
328 301 2123 48 4360 7160
------------------- ------------------- --------------------- -------------------------- -------------------- -------------------
- peaks_classification_* - файл в котором записаны результаты пересечения всех моделей без учёта пересечения сайтов, т.е. результаты приведены исходя из того присутствуют какие сайты в целом есть в пике.
Пример (для трех моделей):
------ ------ -------- ---------------- ------------------ ------------------- ----------------------------- -------------------
pwm bamm sitega bamm\_and\_pwm pwm\_and\_sitega bamm\_and\_sitega bamm\_and\_pwm\_and\_sitega number\_of\_peaks
1793 2494 4956 6403 459 709 1716 46458
------ ------ -------- ---------------- ------------------ ------------------- ----------------------------- -------------------
Публикации связанные с MultiDeNA
-
Tsukanov, A. V., Mironova, V. V., & Levitsky, V. G. (2022). Motif models proposing independent and interdependent impacts of nucleotides are related to high and low affinity transcription factor binding sites in Arabidopsis. Frontiers in plant science, 13, 938545.
-
Tsukanov, A. V., Levitsky, V. G., & Merkulova, T. I. (2021). Application of alternative de novo motif recognition models for analysis of structural heterogeneity of transcription factor binding sites: a case study of FOXA2 binding sites. Vavilovskii zhurnal genetiki i selektsii, 25(1), 7.