Информационно-программный модуль ESDEG
ВВЕДЕНИЕ. Изучение генных регуляторных сетей и механизмов контроля транскрипции представляет собой одну из ключевых задач современной молекулярной биологии. Такие исследования важны для понимания патогенеза заболеваний и поиска новых терапевтических мишеней. Критическую актуальность в этих исследованиях приобретает анализ уровня транскрипции генов, который позволяет выявить молекулярные ответ клеток на различные внешние стимулы или внутренние перестройки. На сегодняшний день технология RNA-seq остается «золотым стандартом» для изучения транскриптома, обеспечивая идентификацию дифференциально экспрессируемых генов (ДЭГ). Однако поиск ДЭГ является лишь первым этапом анализа. Для реконструкции регуляторных каскадов необходимо установить, какие именно транскрипционные факторы (ТФ) были задействованы в наблюдаемом изменении уровня транскрипции. ТФ связываются в регуляторных участках генома со специфическими короткими последовательностями нуклеотидов — сайтами связывания транскрипционных факторов (ССТФ), обеспечивая тем самым активацию или репрессию транскрипции. Для каждого ТФ существует набор ССТФ, который можно представить в виде определенного паттерна — мотива. Важной задачей для анализа ДЭГ является поиск ТФ, чьи мотивы статистически значимо обогащены в промоторах. Для решения этой задачи был разработан информационно-программный модуль (ИПМ) ESDEG (Enrichment of Sites in DEGs). ИПМ ESDEG с помощью метода Монте-Карло оценивает статистическую значимость обогащения мотивов, взятых из баз HOCOMOCO иJASPAR, в промоторах ДЭГ. Важная особенность этого модуля -интеграция иерархической классификации ТФ — TFClass, что позволяет учитывать семейства и подсемейства ДНК-связывающих доменов, а также использовать данны RNA-seq для фильтрации ТФ по уровню их экспрессии. ИСПОЛЬЗУЕМЫЕ МЕТОДЫ И АЛГОРИТМЫ.
- Распознавание ССТФ в промоторах генов с использованием позиционных весовых матриц (PWM).
- Использование множественных порогов распознавания на основе ожидаемых частот встречаемости мотивов (ERR — Expected Recognition Rate).
- Для надёжной оценки статистической значимости обогащени выборок мотивами используется метод Монте-Карло, позволяющий получать эмпирическое распределение обогащения по негативной выборке и затем сравнить наблюдаемые значения с этим распределением.
- Объединение p-значений для множественных порогов с использованием метода Бонферрони.
- FDR-коррекция для множественного тестирования различных мотивов. ФУНКЦИИ И НАЗНАЧЕНИЕ ИПМ. ИПМ ESDEG предназначен для автоматизированной идентификации ТФ, участвующих в регуляции экспрессии генов, на основе анализа промоторных последовательностей дифференциально экспрессирующихся генов. ИПМ позволяет находить ТФ с обогащёнными мотивами и учитывать уровень экспрессии кодирующих их генов уточнения биологически значимых регуляторов, что особенно важно при исследовании молекулярных механизмов заболеваний и определения новых терапевтических мишеней. БЛОК-СХЕМА ИПМ. Общая схема работы ИПМ ESDEG приведена на рисунке Ж.39.
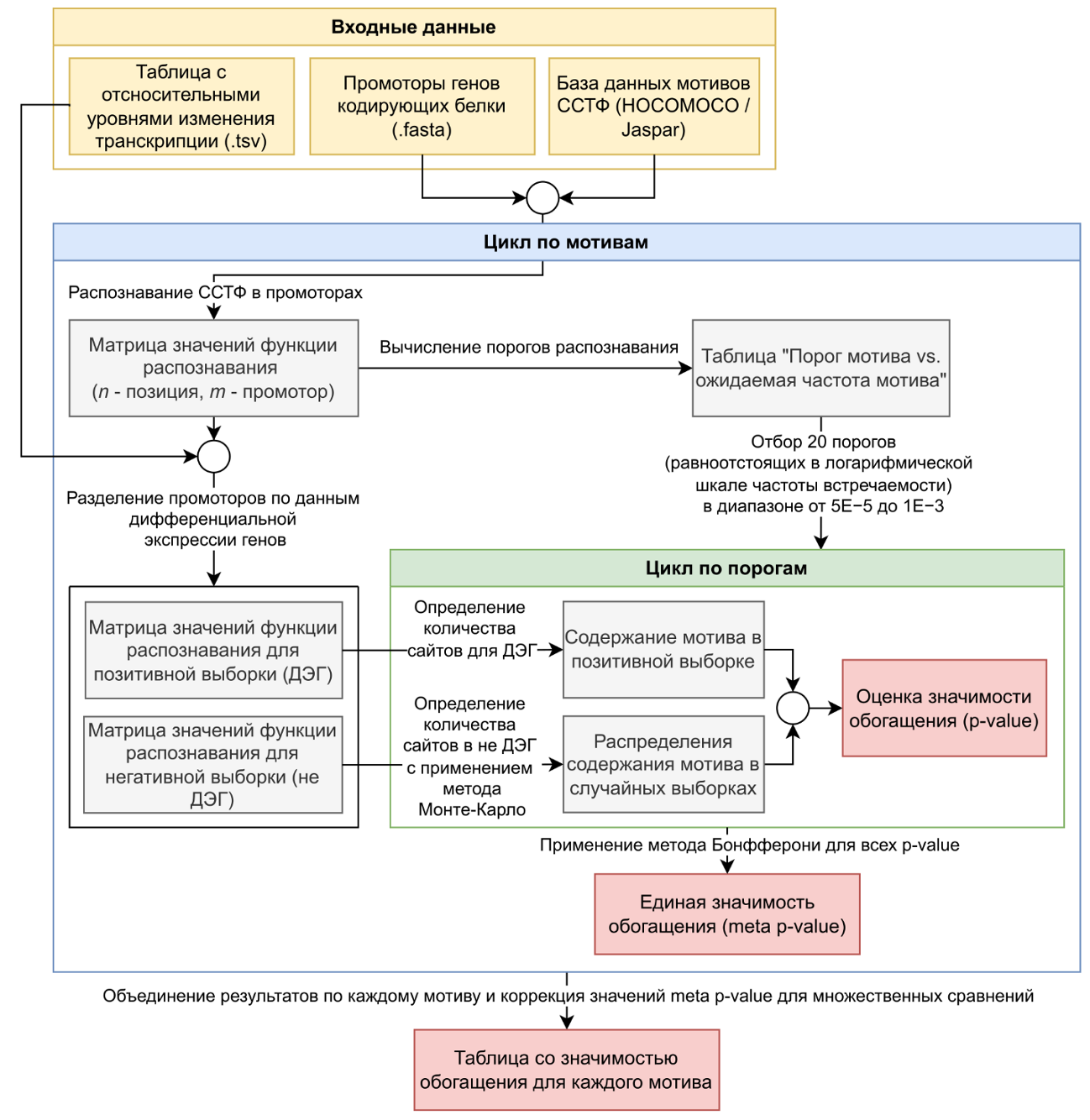
Рисунок Ж.39 – Схема ИПМ ESDEG для анализа данных RNA-seq. Жёлтый цвет – входные данные, серый – промежуточные данные расчётов, формирующихся в процессе работы ИПМ, зелёный/голубой – отдельные блоки/части блоков, розовый – выходные данные (результаты) отдельных блоков. ИПМ ESDEG имеет два блока (один вложен в другой). Во внешнем блоке (голубой цвет) осуществляется подготовка данных по каждому мотиву: распознавание ССТФ, вычисление порогов распознавания и разделение промоторов. Во внутреннем блоке (зеленый цвет) осуществляется оценка значимости обогащения по нескольким порогам, а результатом работы блока является величина meta-p-value, вычесленная модифицированным методом Бонфэрони, описаным в работе (Cinar & Viechtbauer 2022). Итогом работы внешнего блока является объединение величин всех полученных meta-p-value по всем мотивам в сводную таблицу
На первом этапе для каждого мотива выполняется полное сканирование всех позиций в каждом промоторе, т. е. для каждой позиции вычисляется функция распознавания. Итогом является матрица оценок, где индекс n обозначает промотор гена (для N генов), а индекс m соответствует позиции сканирования. На втором этапе на основе полученной матрицы составляется таблица порогов распознавания, включающая значения порога функции распознавания мотива и ожидаемые частоты мотива для полногеномной выборки промоторов. Из полученной таблицы выделяются двадцать порогов, равномерно распределённых в логарифмической шкале ERR с диапазоном от 5×10⁻⁵ до 10⁻³, что позволяет получить гибкое управление чувствительностью оценки обогащения мотива. На третьем этапе промоторы всех генов разделяются на «промоторы ДЭГ» (позитивная выборка) и «промоторы не ДЭГ» (негативная выборка). «промоторы ДЭГ» определяются по значимости изменения транскрипции с учётом множественных сравнений (adjusted p-value ≤ 0.05) и кратности изменения уровня транскрипции (|Log₂(Fold Change)| ≥ 1). Для «промоторов не ДЭГ» нет значимого изменения экспрессии (adjusted p-value > 0.05), а кратность изменения удовлетворяет условию |Log₂(Fold Change)| ≤ 0.32. Следующий этап является основным, на нем происходит оценка обогащения мотива. Предварительно для порога на позитивной выборке вычисляется обогащение мотива одним из двух возможных способов. Первый способ — это вычисление доли промоторов, содержащих хотя бы один предсказанный сайт связывания транскрипционного фактора, второй — оценка доли предсказанных сайтов, представляющая собой отношение общего числа обнаруженных сайтов к общему числу протестированных позиций. Выбор метрики зависит от постановки биологической задачи: первый вариант метрики лучше подходит для выявления транскрипционных факторов, контролирующих общий набор генов, второй — для обнаружения транскрипционных факторов с множественными сайтами на одном промоторе. Далее для оценки статистической значимости обогащения применяется метод Монте-Карло, который позволяет получить эмпирическое распределение величины обогащения мотива на негативной выборке и затем сравнить наблюдаемое значение обогащения с полученным распределением. Процедура состоит из нескольких последовательных шагов. Сначала вычисляется наблюдаемое значение обогащения (Obs) позитивной выборки. Затем для большого числа итераций K (обычно K > 1000) формируется случайная выборка промоторов, на которой также вычисляется обогащение (ExpK). Случайная выборка формируется из негативной выборки путём случайного отбора списка промоторов, равного по объему и распределению содержания G/C с позитивной выборкой. На основе начений ExpK для всех итераций строится эмпирическое распределение (оно считается нормальным), по нему рассчитываются среднее значение (μ) и стандартное отклонение (σ). Далее вычисляется Z-score и рассчитывается соответствующее p-value. Для учёта множественного тестирования по двадцати порогам применяется поправка Бонферрони для вычисления единого meta p-value. Затем для учёта множественного тестирования по большому числу мотивов применяется дополнительная коррекция методом Бенджамини–Хохберга. Каждому обнаруженному обогащённому мотиву автоматически присваиваются атрибуты из системы TFClass: класс и семейство, которые отражают эволюционные и структурные отношения между ТФ. Отдельным анализом (нет на схеме), который может осуществлять ESDEG является фильтрация ТФ с обогащенными мотивами, на основе уровня транскрипции генов соответствующих ТФ. Это позволяет из большого числа потенциальных ТФ-регуляторов выделить именно те, которые скорее всего были задействованы в регуляции. Для этого анализа требуется выходные данные ESDEG по обогащению мотивов ТФ, таблица с уровнями транскрипции генов, а также геномная аннотация. Фильтрация сводится к отбору только тех ТФ уровень транскрипции которых выше заданного порога (например, TPM 2).