Конвейер обработки данных по дфференциальной экспрессии RNA-seq от сырых данных до результирующих таблиц диффренциально экспрессирующихся генов (ДЭГ)
Конвейер, предназначен для обработки данных RNA-seq для получения итоговых данных - таблиц диффренциально экспрессирующихся генов (ДЭГ). Конвейер состоит из модуля предобработки сырых данных – Cutadapt, на котором проводится фильтрация чтений с низким качеством, обрезание концов чтений с низким качеством и содержащих очистка прочтений от последовательностей адаптеров. Далее очищенные чтения поступают на вход программы STAR для картирования на референсный геном. Референсный геном также должен быть подан на вход программы STAR в индексированном этой программой виде, либо индексирование может быть выполнено для предварительно загруженного пользовательского генома. На выходе программы могут быть получены файлы формата BAM (бинарный эквивалент формата SAM, Sequence Alignment/Map). Относительная активность каждого гена определяется согласно покрытию этого гена нуклеотидными прочтениями на референсном геноме после картирования каждой библиотеки. Файлы с этими данными также доступны на выходе программы STAR или могут быть получены из файлов формата BAM другими программами для расчета покрытия. На последнем этапе программа DeSeq2, используется для сравнения полученные данные по покрытию аннотированных генов между состоянием 1 и состоянием 2, результатом является таблица диффренциально экспрессирующихся генов (ДЭГ). На предварительном этапе может быть проведена проверка качества данных РНК-сек с помощью модуля FastQC. На рисунке 1 приведена блок-схема конвейера
Описание входных данных
На вход конвейера подаются сырые данные, полученные при секвенировании RNA-seq - файлы формата fastq.
Описание выходных данных
Выходные данные содержат таблицы диффренциально экспрессирующихся генов, значения уровня изменения экспрессии Log2fold и показатели значимости p adjusted для каждого гена
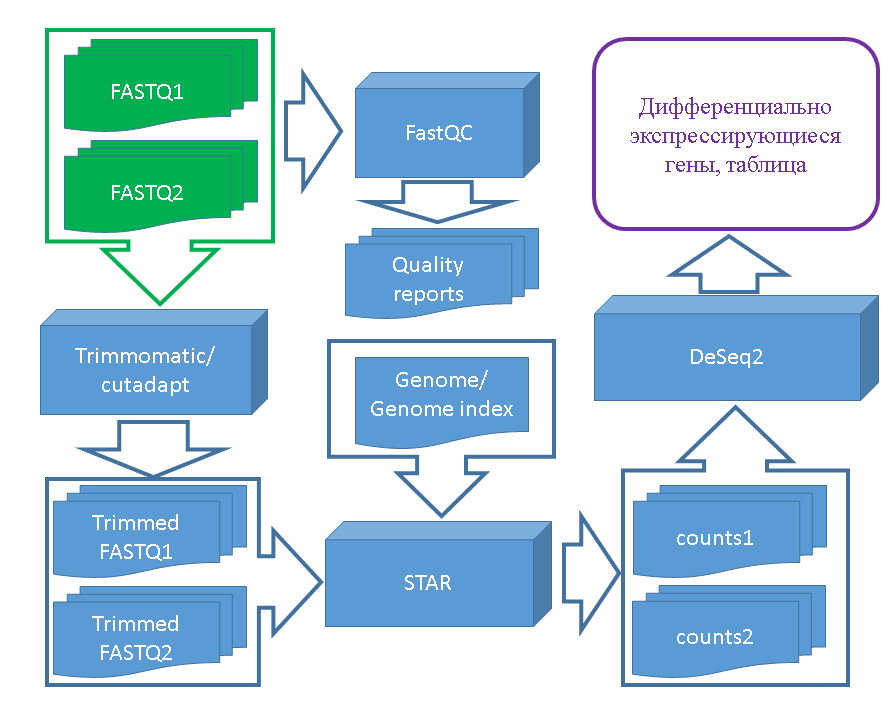
Рисунок 1 Блок-схема для обработки данных RNA-seq для получения таблиц диффренциально экспрессирующихся генов между данными для условий «1» и «2». Зеленым цветом отмечены входные данные - файлы формата FASTQ для условий 1 и 2, полученные при секвенировании RNA-seq. Очищенные чтения соответствуют блокам «Trimmed FASTQ». Данные с покрытием генов прочтениями обозначены как «counts». Три используемых программных компоненты обозначены параллелепипедами. Движение данных в конвейере обозначены стрелками. Результирующие данные – таблица ДЭГ отмечена фиолетовым.