Программный модуль «CROP GENE»
Модуль «CROP GENE» предназначен для решения задач, ориентированных на сельское хозяйство и биотехнологии, интегрирующего методы биоинформатики, анализ больших генетических данных и технологии искусственного интеллекта. Система включает блоки биоинформатического анализа данных: анализ вариаций генов, сборка геномов и транскриптомов, аннотация генов и белков. Программный комплекс CropGene включает программные пакеты, представленные на рисунке 1.
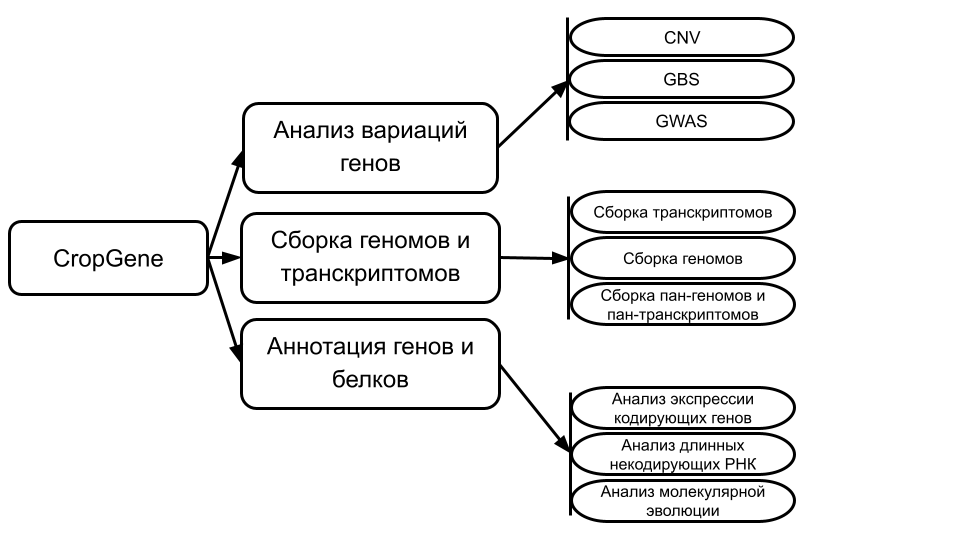
Рисунок 1. Схема программного комплекса CropGene с указанием основных блоков анализа (скругленные прямоугольники в центре) и конкретных решаемых задач (овалы справа).
Структура программного комплекса включает следующие блоки для решения задач. Программный модуль анализа полногеномных ассоциаций.
Этот модуль реализует следующие этапы анализа:
• Анализ данных фенотипирования. Обработка данных фенотипирования производится с использованием пакетов R, pastecs, psych.
• Обработка данных генотипирования. Направлен на процессинг данных генотипирования, полученных методом генотипирования на микрочипах и методом GBS. Обработка включает в себя проверку качества сырых прочтений, картирование на референсный геном с помощью BWA-MEM и поиск полиморфизмов с использованием vcftools. Варианты, определенные вышеуказанными методами генотипирования, фильтруют по качеству, частоте минорного аллеля, гетерозиготности и количеству пропущенных данных. Этот этап осуществляется инструментом bcftools. Для восстановления пропущенных данных генотипирования используют BEAGLE 5.2.
• Полногеномный анализ ассоциаций. На данном этапе осуществляется непосредственно полногеномный анализ ассоциаций, реализуемый на языке программирования R при помощи функций пакета «GAPIT3».
• Приоритизация генов в выявленных локусах. Модуль полногеномного анализа ассоциаций направлен на выявление генов-кандидатов, связанных с интересующими признаками. Первым делом, с использованием функций пакета R «genetics» определяются границы локусов, которые включают в себя значимо ассоциированные с фенотипом варианты. Далее, основываясь на опубликованных данных по экспрессии генов у исследуемого организма и на ресурсах платформы Knetminer, производят приоритизацию генов среди обнаруженных локусов.
Программный модуль анализа CNV.
Этот модуль направлен на решение задач по оценке и анализу вариаций количества копий в геноме. Он реализует несколько этапов анализа:
• Наборы сырых прочтений фильтруются по качеству и длине с помощью программы fastp Далее фильтрованные и обработанные наборы прочтений картируются на референсный геном картофеля с помощью программы BWA. Дупликаты в картированных прочтениях маркируются, удаляются, после чего происходит сортировка и индексирование прочтений с помощью программы.
• Полученные файлы формата BAM используются как входные данные в программе CNVpytor. Вариации количества копий выявлялись на всех хромосомах референсного генома. Выявленные CNV фильтруются следующим образом: длина более 1 т.п.н., p-value < 0.01, q0 < 50% и pN < 50%. Для сопоставления выявленных CNV с генами референсного генома используется R пакет intansv.
• Для последующей обработки список CNVs был представлен в виде матрицы, в которой строки соответствуют конкретному генотипу, а столбцы — гену референсного генома. Каждый элемент матрицы представлен в трех вариантах: +1 (потенциальная дупилкация), -1 (потенциальная делеция) и 0 (отсутствие значимого CNV). Далее проводится анализ главных компонент (PCA) с помощью пакета Scikit-learn v1.1.2, что позволяет оценить генетическое разнообразие.
Биоинформатический конвейер GBS-DP.
Этот программный модуль направлен на анализ данных полученных методом GBS состоит из трех основных этапов:
• Предобработка данных включает проверку качества сырых прочтений FastQC, удаление адаптеров fastp и построение индекса референсного генома.
• Поиск полиморфизмов состоит из картирования предобработанных прочтений на референсный геном Bwa-Mem2, сортировки картированных прочтений Samtools и поиска однонуклеотидных полиморфизмов Bcftools.
• Анализ генетического разнообразия разделяется на два варианта обработки данных: если полученные данные превышают занимаемый объем памяти в 1 Тб и если полученные данные не превышают занимаемый объем памяти в 1 Тб. Выбор соответствующей опции осуществляется автоматически и связан с увеличенной нагрузкой на оперативную память компьютера при работе с большими данными. Анализ главных компонент, отфильтрованных SNP, применяется пакет R – SNPrelate, для построения филогенетического дерева – тоже пакет SNPrelate.
Программный модуль по реконструкции транскриптома.
Этот модуль реализует следующие этапы анализа: • Непосредственно сборка последовательностей контигов из прочтений библиотек RNA-seq. На этой стадии используются программы: Trinity, Trans-ABySS, rnaSpades.
• Объединение полученных наборов контигов и удаление избыточности программой tr2aacds.pl из конвейера EvidentialGene
• Оценка качества полученных последовательностей; программа BUSCO используется для определения полноты транскриптома; программа kallisto показывает, насколько полно исходные библиотеки прочтений были использованы для реконструкции транскриптома; rnaQUAST оценивает различные метрики качества полученного транскриптома, в том числе наличие гомологии с последовательностью генома организма, или генома близкородственного организма, в случае работы с немодельным видом.
Программный модуль реконструкции и анализа пангенома.
Этот модуль реализует следующие шаги анализа:
• Реконструкция каждого генома на основе парных коротких прочтений с помощью геномного сборщика MaSuRCA.
• Маскирование мобильных генетических элементов с помощью RepeatMasker и дальнейшая de novo аннотация реконструированных маскированных геномов с дальнейшей трансляцией открытых рамок считывания с помощью программы AUGUSTUS.
• Выявление ортологических групп в наборе аминокислотных последовательностях, полученных на основе открытых рамок считывания, с помощью OrthoFinder.
Программный модуль оценки экспрессии генов.
В данном модуле оценка экспрессии генов может проводиться как на основе референсного генома, так и на основе транскриптома, реконструированного de novo:
• Для подсчёта экспрессии генов референсного генома проводится выравнивание прочтений библиотек RNA-seq на последовательность генома с помощью программы Dart. Далее используется разметка генома с позициями известных генов для подсчёта количества прочтений, картированных на каждый ген, с помощью программы featureCounts.
• Для оценки экспрессии транскриптов из реконструированного ранее транскриптома используется программа kallisto, которая проводит т.н. псевдовыравнивания прочтений, чтобы определить, к какому транскрипту они принадлежат, на основании чего далее подсчитываются уровни экспрессии
Биоинформатический конвейер ICAnnoLncRNA.
Этот модуль, направлен на выявление и аннотацию днРНК, реализует три этапа обработки транскриптомных последовательностей:
• Контроль качества. Данный этап включает две операции: построение индексного файла для геномной последовательности программой gmap и обучение модели распознавания днРНК программой LncFinder v1.1.4.
• Идентификация днРНК. Данный блок состоит из трех этапов: (1) предсказание кандидатов в днРНК из входного набора транскриптов с помощью метода LncFinder; (2) фильтрация полученных последовательностей-кандидатов на основе идентификации трансмембранных сегментов в ОРС; (3) выравнивание фильтрованных последовательностей-кандидатов днРНК на референсный геном.
• Анализ пан-транскриптомов. Аннотация включает, определение типов последовательностей днРНК по выравниванию на гены кодирующие белок, выявление консервативных днРНК, анализ структурных особенностей днРНК и их экспрессии.
Программный модуль анализа эволюции белков OrthoDOM.
Модуль реализует четыре ключевых этапа анализа белковых последовательностей:
• На первом проводится валидация входных данных и проверка наличия функциональных доменов, заданных пользователем у референсных белков.
• На втором этапе проверяется наличие ключевых доменов в референсных последовательностях
• На третьем — выполняется работа программы Orthofinder для исследуемых протеомов.
• На четвертом производится проверка выявленных ортологов по наличию в их последовательности наборов заданных доменов.
1. Предобработка данных включает проверку качества сырых прочтений генома, удаление адаптеров и построение индекса референсного генома.
2. Поиск полиморфизмов состоит из картирования предобработанных прочтений на референсный геном, сортировки картированных прочтений и поиска однонуклеотидных полиморфизмов.
3. Анализ генетического разнообразия разделяется на два варианта обработки данных: (1) если полученные данные превышают занимаемый объем памяти в 1 Тб, (2) если полученные данные не превышают занимаемый объем памяти в 1 Тб.
Описание входных данных
Входные данные для сырых прочтений – в формате FASTQ, для нуклеотидных и белковых последовательностей в формате FASTA. Аннотация генома в формате GFF.
Описание выходных данных
Выходные данные в различных форматах: FASTA (последовательности транскриптов, Генов и белков), результаты поиска полиморфизмов разделяются по хромосомам в формате VCF, переформатированный формат результатов поиска полиморфизмов, GDS формат, графическая информация в форматах png, прочие данные в формате tsv, филогенетические деревья в формате NPH (newick).