Инструмент SiteGA
Ссылка на репозиторий
https://github.com/parthian-sterlet/sitega
Предназначение программного модуля “SiteGA”
Модель SiteGA реализует подход (Levitsky et al., 2007) для поиска мотивов de novo в одном наборе данных ChIP-seq (пики ChIP-seq) (Tsukanov et al., 2022). SiteGA использует генетический алгоритм, который оценивает мотивы как набор частот взаимозависимых локально позиционированных динуклеотидов (ЛПД). Каждый ЛПД задан типом динуклеотида (один из 16) и начальной и конечной позициями [a, b], каждый из которых располагаются в пределах сайта заданной длины L, 1 ≤ a ≤ L, 1 ≤ b ≤ L. Здесь L – длина мотива в парах нуклеотидов, параметр модели мотива SiteGA. Позиции двух ЛПД одного типа не могут пересекаться. Подход SiteGA моделирует зависимости нуклеотидного контекста внутри сайтов связывания транскрипционных факторов (СС ТФ). Такой подход существенно отличается от традиционной модели позиционной весовой матрицы (ПВМ), которая оценивает сайты по суммам аддитивных вкладов частот нуклеотидов из всех позиций. Подход ПВМ игнорирует любые зависимости между различными позициями внутри ССТФ.
Для заданного набора из M последовательностей позитивной выборки (пиков) функция приспособленности генетического алгоритма F(X) = D(X) * E(X) оценивает выравнивание X = {x(1), x(2), …, x(M)} из M лучших по пикам предсказанных сайтов в модели SiteGA. Первый фактор D(X) отражает зависимости позиций внутри выравнивания с помощью линейного дискриминантного анализа (Levitsky et al. 2007) с помощью метрики «Расстояние Махаланобиса», зависящей от набора из N частот ЛПД. Число ЛПД (NLPD) является параметром модели мотива SiteGA. Второй фактор E(X) означает среднее обогащение k-меров в выравнивании предсказанных сайтов (Tsukanov et al., 2022). Программа индексирует все возможные позиции сайтов в позитивной выборке с весами, равными логарифмам обогащения k-меров при сравнении наборов последовательностей позитивной и негативной выборок. Последовательности негативной выборки готовятся с помощью модуля AntiNoise (Raditsa et al., 2024); для каждой последовательности позитивной выборки случайно из генома выбирается несколько негативных последовательностей, используется для этого подбора только доля в последовательности нуклеотидов A/T и её длина.
Модуль SiteGA использует в работе функционал модуля AntiNoise для подготовки выборке негативных последовательностей ДНК по выборке позитивных последовательностей ДНК. Функционал модуля SiteGA (обучение модели мотива SiteGA, оценка её точности, распознавание СС ТФ) используется в модуле MultiDeNa, реализующем единообразное применение разных моделей мотива СС ТФ.
Коды и адрес программного модуля
Исходный код, конвейерные скрипты и примеры их запуска доступны по адресу https://github.com/parthian-sterlet/sitega
Описание функционала программного модуля “SiteGA”
Общая схема конвейера SiteGA представлена на рисунке.
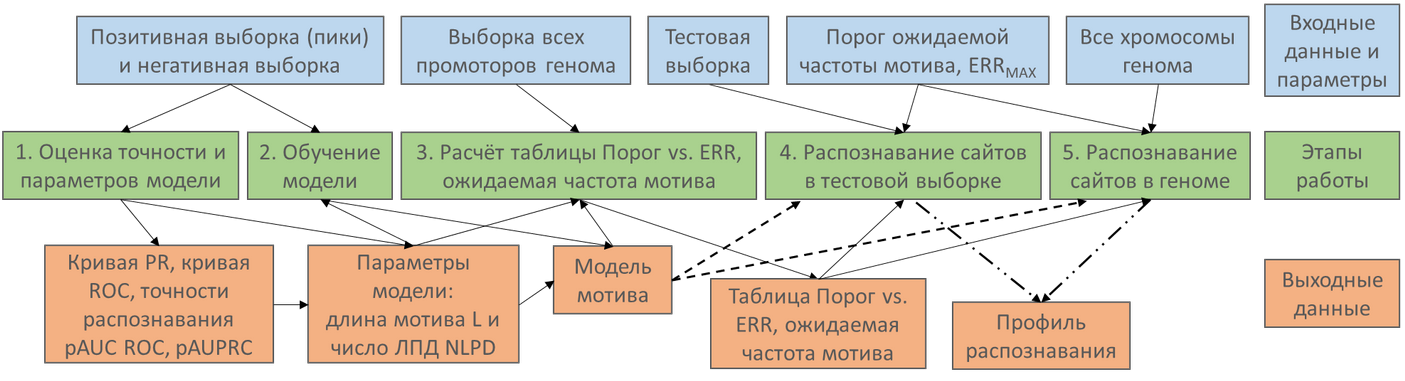
Конвейер SiteGA содержит следующие этапы работы.
-
Оценка точности и параметров модели мотива. Производится путём перекрёстной проверки (двукратного разделения выборки на две подвыборки обучение/контроль, попеременно последовательности с чётными/нечётными номерами берутся в подвыборки обучение/контроль). Ранее пики отсортированы по качеству, так как они является результатом единообразной первичной обработки (Kolmykov et al. 2021). Запуск программы оценки точности для заданной длины мотива L выдаёт значения метрик оценки точности (1) pAUC ROC (partial area under ROC curve, частичная площадь под кривой ROC, Tsukanov et al., 2022) и (2) pAUPRC (partial area under PR curve, частичная площадь под кривой PR, Levitsky et al., 2024) для каждого целого значения из заданного диапазона числа ЛПД в модели от минимального NLPDMIN до максимального NLPDMAX. Термин ROC кривая (Receiver Operating Characteristic curve) означает «Рабочая Характеристика Приемника». Эта стандартная кривая, построение которой используется для оценки точности бинарного классификатора. Кривая ROC показывает зависимость доли предсказанных последовательностей позитивной выборки (ось Y кривой ROC) от частоты мотива в негативной выборке (ось X кривой ROC, Tsukanov et al., 2022). Кривая PR (Precision-Recall, Davis, Goadrich, 2006) – это альтернатива более популярной кривой ROC. Мера Precision (ось Y кривой PR) равна отношению доли предсказанных последовательности позитивной выборки к сумме долей предсказанных последовательностей позитивной и негативной выборок, мера Recall равна доле предсказанных последовательностей позитивной выборки (ось X кривой PR). Для вычисления обеих метрик точности pAUC ROC и pAUPRC важным параметром является максимальная ожидаемая частота мотива (ERRMAX, Maximal Expected Recognition Rate), доля нуклеотидных позиций (пар оснований), содержащих предсказанные сайты. При расчёте меры pAUPRC также учитывается ожидаемое значение меры Precision, равное 0.5, что означает полное отсутствие нуклеотидной специфичности связывания ТФ с ДНК (Saito, Rehmsmeier, 2015; Levitsky et al., 2024).
-
Обучение модели мотива по заданным параметрам L (длина мотива), диапазону значений NLPD (чисел ЛПД в модели). Результатом расчётов являются параметры длины мотива L и числа ЛПД NLPD, соответствующие максимальному значению оценки точности pAUPRC
-
Расчёт таблицы, «Порог функции распознавания vs. Ожидаемая частота мотива в выборке всех промоторов генома» по заданной модели мотива (Threshold vs. ERR, ERR = Expected Recognition Rate, ожидаемая частота мотива)
-
Распознавание сайтов в выборке тестовых последовательностей с помощью модели мотива и выбранным порогом модели
-
Распознавание сайтов во всём геноме с помощью модели мотива и выбранным порогом модели
Алгоритмическая реализация
Каждый из пяти этапов реализован в виде программы на языке с++. Конвейеры запуска, сочетающие один или несколько модулей реализованы на языке perl
Описание входных данных
На вход конвейера подаются
- путь к исполняемым файлам,
- путь к папке с входными данными (позитивной и негативной выборками в формате фаста),
- путь к папке с выходными данными.
- Файл с полным путём к нему, выборка всех промоторов генома.
Параметры запуска
Списки аргументов командной строки для каждого этапа работы приведены ниже.
-
Оценка точности и параметров модели мотива
- путь к файлам в формате фаста с наборами последовательностей позитивной и негативной выборок (последний символ пути должен быть ‘/’ и ‘' для Linux и Windows OS, соответственно)
- файл в формате фаста, набор последовательностей позитивной выборки
- файл в формате фаста, набор последовательностей негативной выборки
- целое число, максимальная длина одного ЛПД (значение по умолчанию 6)
- целое число, минимальное количество ЛПД (значение по умолчанию от 20 до 50)
- целое число, шаг числа ЛПД (значение по умолчанию 1)
- целое число, длина мотива в парах нуклеотидов (значения по умолчанию от 8 до 12)
- вещественное число (формат double), пороговая ожидаемая частота мотива, \(ERR_{MAX}\) (значение по умолчанию 0.01)
- целое число, длина k-мера для учёта обогащения k-меров между позитивной и негативной выборками, значение по умолчанию 6, берутся частоты гексамеров.
- путь к выходным файлам (последний символ пути должен быть ‘/’ и ‘' для Linux и Windows OS, соответственно)
- целое число, максимальная длина пика (по умолчанию 3000)
- лог файл выходной.
-
Обучение модели мотива по заданным параметрам L, NLPD.
- путь к файлам в формате фаста с наборами последовательностей позитивной и негативной выборок (последний символ пути должен быть ‘/’ и ‘' для Linux и Windows OS, соответственно)
- файл в формате фаста, набор последовательностей позитивной выборки
- файл в формате фаста, набор последовательностей негативной выборки
- целое число, максимальная длина одного ЛПД (значение по умолчанию 6)
- целое число, число ЛПД
- целое число, длина мотива в парах нуклеотидов
- целое число, длина k-мера для учёта обогащения k-меров между позитивной и негативной выборками, значение по умолчанию 6, берутся частоты гексамеров.
- путь к выходным файлам (последний символ пути должен быть ‘/’ и ‘' для Linux и Windows OS, соответственно)
- целое число, максимальная длина пика (по умолчанию 3000)
- лог файл выходной
-
Расчёт таблицы «Порог функции распознавания vs. Ожидаемая частота мотива в выборке всех промоторов генома» по заданной модели мотива.
- путь к файлу в формате фаста, выборка всех промоторов генома (последний символ пути должен быть ‘/’ и ‘' для Linux и Windows OS, соответственно)
- файл sitega_matrix_file = входной файл модели SiteGA
- файл в формате фаста, выборка всех промоторов генома
- выходной файл в текстовом формате, таблица Threshold vs. ERR, «Порог функции распознавания vs. Ожидаемая частота мотива в выборке всех промоторов генома»
- выходной файл в бинарном (двоичном формате), модель SiteGA в виде матрицы и таблица Threshold vs. ERR, «Порог функции распознавания vs. Ожидаемая частота мотива в выборке всех промоторов генома»
- число в формате double, pvalue_large = максимальная ожидаемая частота мотива \(ERR_{MAX}\) (значение по умолчанию 0.01)
- число в формате double dpvalue = значение шага для уплотнения значений ожидаемой частоты мотива ERR в таблице Threshold vs. ERR, значение по умолчанию 0.0000005 = 5E-7 подразумевает умеренное уплотнение
-
Распознавание сайтов в выборке тестовых последовательностей с помощью модели мотива и выбранным порогом модели
- файл тестовой выборки в формате фаста с указанием пути к нему
- sitega_matrix_file, файл модели SiteGA
- целое число, режим работы, site_description_mode = 0 или 1. 0 означает режим по умолчанию, 1 означает вычисление частот всех ЛПД для всех последовательностей (опция используется для описания модели SiteGA только для файла в формате фаста, который был на обучении, позитивной выборки)
- вещественное число (формат double), порог для ожидаемой частоты мотива модели SiteGA, \(ERR_{MAX}\). Выбирается по рассчитанной заранее таблице Threshold vs. ERR, (значение по умолчанию от 5E-5 до 0.01)
- входной файл в текстовом формате, Таблица Threshold vs. ERR для выбора порога модели SiteGA
- выходной файл, профиль с распознанными сайтами
-
Распознавание сайтов во всём геноме с помощью модели мотива и выбранным порогом модели
- путь к последовательностям хромосом в формате фаста, это файлы для отдельных хромосом в отдельных файлах, они готовятся путем разбиения фаста-файла целого генома с помощью одной из компонент fasta_muliplefiles.cpp из пакета AntiNoise.
- виды организмов и выпуски геномов (значения hg38, mm10, rn6, zf11, dm6 и ce235; at10, gm21, zm73 и mp61; sc64 и sch294). Животные - человек Homo sapiens hg38, мышь Mus musculus mm10, крыса Rattus norvegicus Rnor_6.0, данио рерио Danio rerio GRCz11, муха Drosophila melanogaster dm6 и круглый червь Caenorhabditis elegans WBcel235; растения - арабидопсис Arabidopsis thaliana TAIR10, соя Glycine max v2. 1, кукуруза Zea mays B73 и печеночница Marchantia polymorpha MpTak v6.1; грибы - пекарские дрожжи Saccharomyces cerevisiae R64-1-1 и делящиеся дрожжи Schizosaccharomyces pombe ASM294v2.
- sitega_matrix_file, файл модели SiteGA
- входной файл в текстовом формате, Таблица Threshold vs. ERR для выбора порога модели SiteGA
- вещественное число (в формате double), порог для ожидаемой частоты мотива модели SiteGA, \(ERR_{MAX}\). Выбирается по рассчитанной заранее таблице Threshold vs. ERR
- выходной файл, профиль с распознанными сайтами
Описание выходных данных
Выходные данные по этапам работы (все файлы в текстовом формате). Приведены примеры выходных файлов для тестовой модели мотива для пиков по данных ChIP-seq PEAKS035299 по ТФ STAT5B, длина мотива 12 нт, диапазон значений ЛПД от 30 до 33 с шагом 1.
- Оценка точности и параметров модели мотива
-
Кривая ROC, позволяет рассчитать меру точности частичная площадь под кривой ROC, (pAUC ROC) – не является способом по умолчанию расчёта точности модели мотива. В колонках представлены: частота предсказанных сайтов в негативной выборке (False Positive Rate) и доля предсказанных пиков (True Positive Rate). Пример (файл ROC_12_33)
0 0 2.20379e-07 0.001000 4.40758e-07 0.002000 8.81516e-07 0.003000 1.32227e-06 0.006000 1.76303e-06 0.013000 2.64455e-06 0.014000 3.52606e-06 0.015000 3.96682e-06 0.016000 4.40758e-06 0.018000 4.84834e-06 0.020000 5.72985e-06 0.022000 6.17061e-06 0.023000 6.61137e-06 0.026000 7.05212e-06 0.029000 7.93364e-06 0.034000 9.25591e-06 0.036000 9.69667e-06 0.038000 1.01374e-05 0.045000 … ```
- Кривая PR, позволяет рассчитать меру точности частичная площадь под кривой PR (pAUPRC) – является способом по умолчанию расчёта точности модели мотива. В колонках представлены: доля предсказанных пиков (Recall) и отношение вероятности предсказания последовательности позитивной выборки к сумме вероятностей предсказания последовательностей позитивной и негативной выборок (Precision), см. (Levitsky et al., 2024). Пример (пример PRC_12_33)
0 1
0.001000 1.000000
0.002000 0.908642
0.003000 0.881790
0.004000 0.868950
0.005000 0.892338
0.006000 0.908642
0.007000 0.896936
0.008000 0.908642
0.009000 0.917960
0.011000 0.931860
0.012000 0.937182
0.013000 0.941733
0.014000 0.920658
0.015000 0.903142
0.016000 0.898383
0.018000 0.899512
0.020000 0.900416
…
-
Сводная таблица по мерам точности, тестовый файл содержит пять колонок: длина мотива, число ЛПД, значение метрики точности pAUC ROC и значение метрики точности pAUPRC. Каждая строка даёт информацию для отдельного числа ЛПД в диапазоне от минимального до максимального, заданного параметрами.
Пример сводной таблицы по мерам точности. В таблице указаны значения длины мотива, числа ЛПД, метрик точности pAUC ROC и pAUPRC.
PEAKS035299_STAT5B_P42232_MACS2.fa 12 30 0.923274 0.660498
PEAKS035299_STAT5B_P42232_MACS2.fa 12 31 0.936624 0.753945
PEAKS035299_STAT5B_P42232_MACS2.fa 12 32 0.923039 0.643664
PEAKS035299_STAT5B_P42232_MACS2.fa 12 33 0.940502 0.75707
- лог файл выходной
- Обучение модели мотива по заданным параметрам L, NLPD
- Модель SiteGA в виде таблицы
Ниже приведён пример весовой матрицы модели SiteGA для набора данных ChIP-seq из базы данных GTRD (Kolmykov et al., 2021), ID PEAKS035299 для ТФ STAT5B. Первый параметр – число ЛПД (LPD count), второй параметр – длина мотива (Model length). Параметры Minimum и Razmah используются для нормировки значения функции распознавания SiteGA, если Score это сумма весов по частотам всех ЛПД, то нормированное функции распознавания, изменяемое в пределах от 0 до 1 равно (Score – Minimum) / Razmah. Далее идёт таблица из пяти колонок и списка ЛПД модели. Для каждого ЛПД модели первая и вторая колонки это его начальная и конечная позиции в пределах длины мотива, третья колонка это вес, четвёртая и пятая номер в алфавите (aa, ac, ag, at, ca … соответствует номерам 0, 1, 2, 3, 4, 5, …) и его тип (aa, ac, ag, at, ca …). В тестируемой последовательности вычисляется частота заданного динуклеотида в районе, опредлённом начальной и конечной позицией и эта частота умножается на её вес, указанный в третьей колонке матрицы SiteGA. Затем, сумма весов по всем ЛПД нормируется как указано выше. (ниже – содержимое файла PEAKS035299_STAT5B_P42232_MACS2)
33 LPD count
12 Model length
0.000000000000 Minimum
2.753926121402 Razmah
0 1 0.036082022180 0 aa
4 4 0.033018104701 0 aa
7 7 0.324865827807 0 aa
8 8 0.091108867582 0 aa
0 1 0.083053548618 1 ac
8 8 0.088927028606 1 ac
4 4 0.034068216735 2 ag
5 5 0.119417820425 2 ag
8 8 0.080218906927 2 ag
8 8 0.075948604149 3 at
2 4 0.096724616572 4 ca
10 10 0.010193897507 4 ca
1 3 0.069136147168 5 cc
0 4 0.248158137134 6 cg
5 5 0.153479195926 6 cg
0 1 0.035965664907 7 ct
2 3 0.078340939375 7 ct
0 0 0.022083392877 8 ga
4 4 0.016058093506 8 ga
6 6 0.369339255420 8 ga
1 2 0.079519577543 9 gc
9 9 0.014321397089 9 gc
5 5 0.108691575162 10 gg
4 4 0.047429189572 11 gt
0 4 0.094698968056 12 ta
0 1 0.067325127522 13 tc
2 3 0.031253247174 13 tc
3 4 0.065538557390 14 tg
5 5 0.055128337598 14 tg
0 0 0.043721465137 15 tt
1 1 0.020338387163 15 tt
2 3 0.043718671193 15 tt
9 9 0.016053332683 15 tt
-
Расчёт таблицы, «Порог функции распознавания vs. Ожидаемая частота мотива в выборке всех промоторов генома» по заданной модели мотива
- Таблица из двух колонок, «Порог функции распознавания vs. Ожидаемая частота мотива в выборке всех промоторов генома»
Пример таблицы для набора данных PEAKS035299, в первой колонке порог функции распознавания модели мотива, во второй колонке частота мотива в логарифмической шкале, \(-Log_{10}(ERR)\), здесь ERR это отношение числа распознанных позиций с сайтами к числу всех возможных позиций сайтов в выборке.
0.408545486866335850 7.29763180083726315
0.404843899729419365 6.25623911567903779
0.403461623088487320 5.97541250610334362
0.402669358198356808 5.80627010700299095
0.401862620552213223 5.6848479441175277
0.401045265605576406 5.59006162473932644
0.400762155911151108 5.51230196582649601
0.400138866302861718 5.44637345211818769
0.399916204764933003 5.38914678195861363
0.399813722207658317 5.33859040851616928
0.399760035951570836 5.29331042705462096
0.399742594470542445 5.2445533573538432
0.399643543417134151 5.20772668939786509
0.399474881612196742 5.17378015987017736
0.399123939874802547 5.14229576337220173
0.398967771061440324 5.11294037001966473
0.398950329580411989 5.08544419643330503
0.398682616722066285 5.05958569770846811
0.398409983821726210 5.03518071110683341
0.398290809741256358 5.01207449182948928
……
- Распознавание сайтов в выборке тестовых последовательностей с помощью модели мотива и выбранного порога модели мотива
Пример основного файла выходных данных (профиля предсказанных сайтов) для модели мотива SiteGA, построенной для набора данных PEAKS035299. В качестве тестируемой выборки взяты все промоторы генов мыши, кодирующих белки (список начинается с генов в начале первой хромосомы). Пример представляет собой список предсказанных сайтов для каждой последовательности. Строки с первым символом ‘>’ означают последовательности из тестовой выборки. Любая другая строка содержит аннотацию распознанного сайта (позиция, значение функции распознавания, цепь ДНК и последовательность сайта)
>chr1 3671499 3673498 - ENSMUSG00000051951.5
1490 0.392829878672666000 - ttcctcgaagct
>chr1 4409242 4411241 - ENSMUSG00000025900.12
1826 0.389714219198197209 - ttctaagaaagg
>chr1 4497355 4499354 - ENSMUSG00000025902.13
1727 0.384870004977334812 - ttcttagaaccc
1730 0.393415806335113694 + ttctaagaaaca
1792 0.396322005815267331 - cgcggcgaagca
>chr1 4785740 4787739 - ENSMUSG00000033845.13
627 0.387544076290433082 + ttcctggaattg
1006 0.384000730600813689 + gacctagaacca
1524 0.398409983821726210 - acgctagaagcg
……
- Распознавание сайтов во всём геноме с помощью модели мотива и выбранного порога модели мотива
Входные данные – набор полноразмерных хромосом полного генома в простом формате, (формат фаста без строки, начинающейся с символа ‘>’), каждая хромосома в отдельном файле.
Формат файла выходных данных (профиля предсказанных сайтов) тот же, что и для описанный выше для тестовой выборки последовательностей.
Список цитированной литературы
1. Davis J., Goadrich M. The relationship between Precision-Recall and ROC curves. In: Proceedings of the 23rd International Conference on Machine Learning. New York: Assoc. for Computing Machinery, 2006, 233-240. doi 10.1145/1143844.1143874
2. Kolmykov S., Yevshin I., Kulyashov M., Sharipov R., Kondrakhin Y., Makeev V.J., Kulakovskiy I.V., Kel A., Kolpakov F. GTRD: an integrated view of transcription regulation. Nucleic Acids Res. 2021; 49(D1), D104-D111.
3. Levitsky, V. G., Ignatieva, E. V., Ananko, E. A., Turnaev, I. I., Merkulova, T. I., Kolchanov, N. A., Hodgman T. C. Effective transcription factor binding site prediction using a combination of optimization, a genetic algorithm and discriminant analysis to capture distant interactions. BMC Bioinformatics 2007, 8, 481.
4. Levitsky V.G., Tsukanov A.V., Merkulova T.I. MetArea software package for analysis of mutually exclusive occurrence in pairs of transcription factor binding site motifs in ChIP-seq data. Vavilovskii Zhurnal Genetiki i Selektsii. 2024, 28(8), 822-833.
5. Raditsa, V. V., Tsukanov, A. V., Bogomolov, A. G., Levitsky, V. G. Genomic background sequences systematically outperform synthetic ones in de novo motif discovery for ChIP-seq data. NAR Genom Bioinform. 2024, 6(3), lqae090.
6. Saito T., Rehmsmeier M. The Precision-Recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One. 2015, 10(3), e0118432.
7. Tsukanov, A. V., Mironova, V. V., Levitsky, V. G. Motif models proposing independent and interdependent impacts of nucleotides are related to high and low affinity transcription factor binding sites in Arabidopsis. Front Plant Sci. 2022, 13, 938545.