Вычислительный конвейер AntiNoise
Ссылка на приложение в Galaxy
https://galaxy.icgbio.ru/root?tool_id=antinoise
Выборки негативных последовательностей ДНК необходимы для проведения анализа обогащения мотивов в выборках последовательностей, в частности, для поиска мотивов de novo в данных массового картирования ChIP-seq (Tsukanov et al., 2022) для определения нуклеотидной специфичности сайтов связывания транскрипционных факторов. Последовательности из референсного генома выбираются в негативную выборку случайным образом, для каждой последовательности позитивной выборки используется для этого подбора только доля в ней нуклеотидов A/T и её длина, что позволяет моделировать ожидаемое содержание неспецифических мотивов, обогащённых в геноме, например, полиА трактов, содержание которых во всех геномах эукариот существенно выше, чем ожидаемое на основе Марковских моделей, построенных по частотам коротких k-меров (Karlin, Burge, 1995).
Описание функционала программного модуля “AntiNoise”
Конвейер AntiNoise предоставляет два способа подготовки выборок негативных последовательностей по заданным выборкам позитивных последовательностей (Raditsa et al., 2024):
- негативные последовательности извлекаются из немаскированного референсного генома,
- негативные последовательности извлекаются из референсного генома с маскированием: доступны опции «Исключение участков из чёрного списка» или «Сохранение участков из белого списка».
- Опция «чёрного списка» исключает определенные геномные локусы из всего эталонного генома, процедура извлечения применяется к оставшимся геномным участкам.
- Опция «белого списка» извлекает негативные последовательности только из определенных заданных участков, все остальные локусы исключаются из анализа.
Коды и адрес программного модуля
Исходный код, конвейерные скрипты и примеры их запуска доступны по адресу https://github.com/parthian-sterlet/antinoise, веб интерфейс доступен по адресу https://denovosea.icgbio.ru/antinoise
Описание функционала программного модуля “ AntiNoise”
Общая схема конвейера AntiNoise представлена на рисунке.
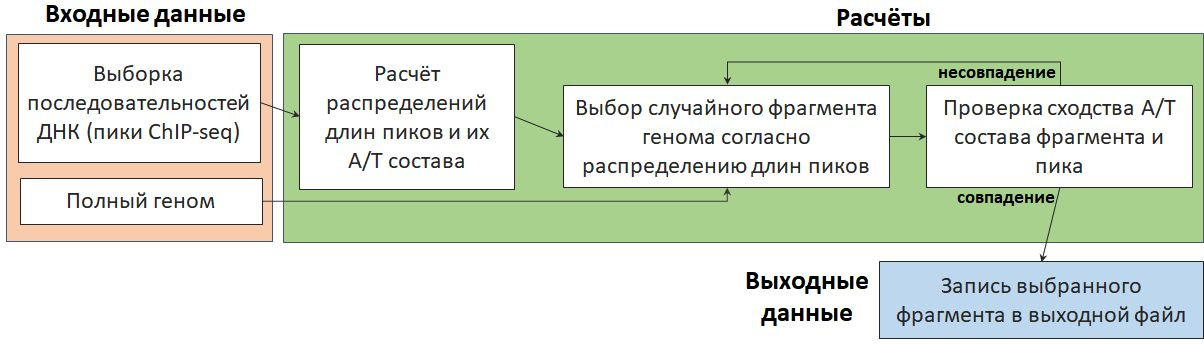
Конвейер AntiNoise
Алгоритмическая реализация
Программа реализована на языке C++. Конвейеры запуска, сочетающие один или несколько модулей реализованы на языке Perl
Описание входных данных
На вход конвейера подаются
- путь к исполняемым файлам,
- путь к референсному геному, маскированному или нет,
- путь к разметке входного файла позитивных последовательностей в формате BED, а также разметки «чёрного» или «белого» списка, если они применяются,
- путь к папке с выходными данными.
Параметры запуска
- Вид и выпуск референсного генома (значения hg38, mm10, rn6, zf11, dm6 и ce235; at10, gm21, zm73 и mp61; sc64 и sch294 означают животных: человека H. sapiens, мышь M. musculus, крысу R. norvegicus, данио рерио Danio rerio, плодовую мушку D. melanogaster и круглого червя Caenorhabditis elegans; и растения: арабидопсис A. thaliana, сою Glycine max, кукурузу Zea mays и маршанцию (печёночный мох) M. polymorpha; грибы: пекарские дрожжи S. cerevisiae R64-1-1 и делящиеся дрожжи S. pombe).
- Необходимое количество негативных последовательностей на одну позитивную последовательность, \(R_{BF}\) (Required Background Fraction). Значение по умолчанию 5.
- Отклонение Delta содержания нуклеотидов A/T в негативной последовательности от такового для позитивной. Значение по умолчанию 0.01.
- Максимальное число попыток \(N_{A}\) найти совпадающие фоновые последовательности в геноме. Если заданное число \(N_{A}\) последних попыток найти хотя бы еще одну негативную последовательность безуспешно, алгоритм завершается. Значение по умолчанию 50000.
- Порог \(F_{MIN}\) для доли полностью обработанных входных последовательностей, позволяющий прекратить вычисления (значение по умолчанию 0.99)
Предобработка входных данных может включать следующие дополнительные этапы:
- Преобразование файла геномной аннотации в формате BED с возможно перекрывающимися по координатам геномными участками в файл в формате BED с неперекрывающимися геномными участками. Эта опция необходима для корректной обработки некоторых видов «чёрных» или «белых» списков, например, промоторов генов.
- Маскирование референсного генома в соответствии с заданной разметкой участков в формате BED. Маскирование с помощью ‘N’ нуклеотидов по выбору выполняется либо
- для всех участков, указанных в BED-файле (опция «чёрный список»), то есть эти участки удаляются из последующего анализа;
- для всех оставшихся частей референсного генома (опция «белый список»), то есть эти только эти участки остаются в последующем анализа.
Список и описание вспомогательных программ, выполняющих эти дополнительные этапы
- Программа area_self_overlap.cpp преобразует файл геномной аннотации в формате BED с возможно перекрывающимися геномными фрагментами в файл в том же формате BED с неперекрывающимися геномными фрагментами.
- Программа bed_chr_mask.cpp маскирует референсные геномные последовательности в простом формате PLAIN (формат фаста без строки, начинающейся с символа ‘>’) в соответствии с заданной входной аннотацией в формате BED. Маскирование с помощью ‘N’ нуклеотидов по выбору выполняется либо (а) для всех фрагментов, перечисленных в BED-файле, либо (б) для всех оставшихся частей референсного генома (опции белого и чёрного списка). Для маскирования референсного генома в формате PLAIN программе необходим файл в формате BED с любыми непересекающимися геномными районами.
- Программа bed_chr_separation.cpp разбивает файл в формате BED на несколько файлов в формате BED так, чтобы в каждом выходном файле были представлены аннотации для одной и той же хромосомы.
- Программа bed_sort.cpp сортирует аннотации ДНК в формате BED в соответствии с именами хромосом и позициями на хромосомах.
- Программа fasta_muliplefiles.cpp разбивает файл в формате FASTA с несколькими последовательностями на несколько файлов так, чтобы каждый выходной файл в формате FASTA соответствовал одной последовательности ДНК (одному участку генома). В частности, эта программа используется для преобразования референсного генома в одном файле FASTA с последовательностями всех хромосом, объединённых вместе, в несколько файлов FASTA с отдельными хромосомами. Геномы в формате FASTA доступны на многих публичных серверах, например, Ensembl, https://www.ensembl.org/.
- Программа fasta_to_plain0.cpp преобразует последовательности ДНК референсного генома из формата FASTA в формат PLAIN. Эта программа делает последовательность референсного генома более удобной для последующего анализа и обработки. В отличие от формата FASTA, (а) формат PLAIN не содержит строк заголовка последовательности с первым символом ‘>’, (б) символ конца строки (’\n’ в Linux или ‘\r\n’ в Windows) означает только конец последовательности ДНК. Поэтому, если файл PLAIN содержит только одну хромосому, то конец строки допускается только в конце файла.
- Программа longext_many.cpp преобразует файл в формате BED в последовательности ДНК в формате FASTA, используя геномные последовательности хромосом в формате PLAIN.
Описание выходных данных
В выходные данные программы включают следующие файлы:
- выборка негативных последовательностей в формате FASTA и её полногеномная разметка в формате BED,
- таблица сравнения содержания нуклеотидов A/T в наборах входных и выходных последовательностей,
- таблица сравнения частот динуклеотидов в наборах входных и выходных последовательностей,
- таблица сравнения (а) частот нуклеотидов A/T во всех отдельных входных последовательностях, которые не достигли требуемого количества найденных выходных последовательностей на одну входную последовательность \(R_{BF}\), и (б) средней частоты нуклеотидов A/T для выборки входных последовательностей.
- таблица сравнения (а) частот динуклеотидов во всех входных последовательностях, не достигших требуемого количества найденных выходных последовательностей на одну входную последовательность \(R_{BF}\), и (б) средние частоты динуклеотидов для выборки входных последовательностей.
Список программ для компиляции и сборки модуля
ОС Линукс
Компиляторы С++ GCC или Intel
Для установки программы необходимо выполнить следующие этапы:
- Скачать программу и выполнить следующие команды в системе Линукс
git clone https://github.com/parthian-sterlet/antinoise
cd antinoise/run
chmod a+x build.sh
- Компиляция всего исходного кода на языке С++
./build.sh
Инструкция по запуску с помощью скриптов на языке Perl, с примерами и тестовых наборов входных данных
- Простой запуск, старт с одного файла полного генома в формате PLAIN
Основная программа конвейера работает с нуклеотидными последовательностями полного генома в простом формате (PLAIN), каждая хромосома в отдельном файле, в котором возможны только символы нуклеотидов. Файл командной строки command_line_simple и perl-скрипт simple.pl позволяют использовать формат BED или FASTA для позитивных последовательностей.
- Запуск без проведения маскирования генома, старт с одного файла полного генома в формате FASTA
Файл командной строки command_line_no_mask и Perl-скрипт no_mask.pl показывают пример применения экстракции геномных негативных последовательностей для тестового набора позитивных последовательностей в формате BED из генома S. cerevisiae. Поиск осуществляется в полноразмерных последовательностях референсного генома. Скрипт начинает работу с одного FASTA-файла, содержащего все хромосомы определенного референсного генома, например. Этот файл должен быть взят в общедоступной базе данных, например, для человека и мыши.
- Запуск с проведением маскирования в геноме участков, занесенных в чёрный список, старт с одного файла полного генома в формате FASTA
Файл командной строки command_line_blacklisted и Perl-скрипт mask.pl показывают пример применения экстракции геномных негативных последовательностей для тестового набора позитивных последовательностей в формате BED из генома S. cerevisiae. В данном примере применяется дополнительный BED-файл с «чёрным списком» участков. В частности, файлы простые повторов и микросателлиты (ди- и тринуклеотидные повторы) (Benson et al., 1999) показывают примеры файлов с возможными перекрываниями участков и без него. Здесь и далее примеры разметок «чёрного» и «белого» списков получены из архива геномного бразуера Калифорнийского Университета Санта Круз, https://genome.ucsc.edu/.
- Запуск с проведением маскирования в геноме участков, занесенных в белый список, старт с одного файла полного генома в формате FASTA
Файл командной строки command_line_whitelisted и Perl-скрипт mask.pl показывают пример применения экстракции геномных негативных последовательностей для тестового набора позитивных последовательностей в формате BED из генома S. cerevisiae. В данном примере применяется дополнительный файл в формате BED с «белым списком» участков. Только эти регионы смогут попасть в выходной файл, все остальные геномные участки будут маскированы и впоследствии исключены из анализа. Регуляторные элементы из экспериментов ChIP-chip (“probes from ChIP-chip experiments for transcription regulatory elements”, Harbison et al., 2004) и консервативные области генома S. cerevisiae с другими видами дрожжей (“conservation - multiz alignment & conservation (7 yeasts)”, Felsenstein and Churchill, 1996; Blanchette et al., 2004) использованы как примеры файлов «белого списка» с возможными перекрываниями участков и без него. Например, промоторные районы генов, кодирующих белки являются перспективным примером для проведения более точного de novo поиска мотивов в данных ChIP-seq.
Примеры результатов для простого запуска (вариант 1, представленный выше), здесь и далее набор пиков эксперимента ChIP-seq (Spivak and Stormo, 2016) для транскрипционного фактора GCN4 пекарских дрожжей S. cerevisiae из базы данных GTRD (ID PEAKS043150, Kolmykov et al., 2021) демонстрирует применение программы.
Входной файл в формате FASTA
>chrXV 748321 748421 + peak900
TCCAAAAAAACCTCTTCTGTACTTTCTTGCTCCTTTTCTAGGTTTTCTAACTCTGTATTTAGGGTCACAATCGTGGATTGTAATTCAGAGTTTTCGGTCTT
>chrIV 290681 290784 + peak869
CCATTGGTTCTTCTATCAAATATATGGCGTTCCAGTTTACCTGTCTATTAGCTATTCCTGCCTTCATCTATTTTCTCATTTTCAGCGTGCATATTAAGACATTA
>chrVII 719433 719533 + peak944
TCACAAGTAAGAAAATACAGGTCATATGACAACAGAGACAAGAGGAAGCCCTCCAAGATAGGCAACAACTTACAAGTAGAGAATGAGGAGGACTATGAAGA
Выходной файл в формате FASTA
>chrX 656028 656129 peak900_1_Mo_0.000000
TTTTTGCTTTCCTCAAATAACTGTATCTACAAATCATTTCATGATTCTTCTAGTCCCCTTTTACTTTTCCTAATCACCTCGCACAACGTACGGTGTGACAT
>chrVII 144242 144346 peak869_1_Mo_0.009615
CGGATGTTTAAAAATTGTTGTCGCCTGGAGAAAGCTAATACTAGTTCTGGCCATATTTTCAACCATTAAACACTACTAATGTAATTACTTGACTTATTTGCTCC
>chrXIII 459550 459651 peak944_1_Mo_0.009901
TCAGGGTCAAATATGATACCCTTTCCATGCAACCTCCATTTGTTAGTCCACTCGATAGCCGTTGATTTTTCATTTTGGCTTTTATTCACTTGAAGCCTTCT
…
Выходной файл в формате BED
chrX 656028 656129
chrVII 144242 144346
chrXIII 459550 459651
…
Доля нуклеотидов A/T, сравнение распределений для позитивной и негативной выборок
A/T content (%): Foreground set vs. Background set 40.000000 80.000000
Foreground Background
40.000000 0.000000 0.000000
42.000000 0.000000 0.000000
44.000000 0.000000 0.060000
46.000000 0.400000 0.320000
48.000000 0.900000 0.780000
50.000000 1.300000 1.380000
52.000000 2.400000 2.460000
54.000000 5.900000 5.020000
56.000000 8.800000 8.600000
58.000000 13.100000 12.440000
60.000000 17.500000 16.400000
62.000000 18.100000 16.740000
64.000000 15.900000 17.280000
66.000000 7.800000 9.540000
68.000000 5.200000 5.840000
70.000000 1.700000 2.100000
72.000000 0.800000 0.700000
74.000000 0.200000 0.300000
76.000000 0.000000 0.040000
78.000000 0.000000 0.000000
80.000000 0.000000 0.000000
Частоты динуклеотидов, сравнение распределений для позитивной и негативной выборок
Dincucleotide frequencies (%): Foreground set vs. Background set
Foreground Background
AA 10.601901 10.204282
AC 5.559336 5.439803
AG 6.254550 5.964092
AT 7.604056 8.422991
CA 7.081099 6.690808
CC 4.107047 4.325556
CG 2.893158 3.218638
CT 6.147484 5.973419
GA 6.442986 6.314698
GC 4.020442 4.077830
GG 3.977140 4.171953
GT 5.550295 5.340636
TA 5.873396 6.807486
TC 6.517694 6.385028
TG 6.900752 6.571180
TT 10.468663 10.091601
…
Список цитированной литературы
1. Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27(2), 573-580.
2. Blanchette, M., Kent, W. J., Riemer, C., Elnitski, L., Smit, A. F., Roskin, K. M., et al. (2004). Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res., 14(4), 708–715.
3. Felsenstein, J., Churchill, G.A. (1996) A Hidden Markov Model approach to variation among sites in rate of evolution. Mol Biol Evol. 13(1), 93-104.
4. Harbison, C. T., Gordon, D. B., Lee, T. I., Rinaldi, N. J., Macisaac, K. D., Danford, T. W., et al. (2004). Transcriptional regulatory code of a eukaryotic genome. Nature, 431(7004), 99–104.
5. Karlin, S., Burge, C. (1995) Dinucleotide relative abundance extremes: a genomic signature. Trends Genet. 11(7), 283-290.
6. Kolmykov, S., Yevshin, I., Kulyashov, M., Sharipov, R., Kondrakhin, Y., Makeev, V. J., Kulakovskiy, I. V., Kel, A., Kolpakov, F. (2021). GTRD: an integrated view of transcription regulation. Nucleic Acids Res. 49, D104–D111.
7. Raditsa, V. V., Tsukanov, A. V., Bogomolov, A. G., Levitsky, V. G. Genomic background sequences systematically outperform synthetic ones in de novo motif discovery for ChIP-seq data. NAR Genom Bioinform. 2024, 6(3), lqae090.
8. Spivak, A. T., Stormo, G. D. (2016). Combinatorial Cis-regulation in Saccharomyces Species. G3 (Bethesda), 6(3), 653–667.
9. Tsukanov, A. V., Mironova, V. V., and Levitsky, V. G. (2022). Motif models proposing independent and interdependent impacts of nucleotides are related to high and low affinity transcription factor binding sites in Arabidopsis. Front. Plant Sci. 13, 938545.