Программный конвейер для предсказания уровней представленности белков в клетках дрожжей на основе анализа контекста их геномов
Ссылка на репозиторий
https://gitea.sysbio.ru/program_providing_fund/YeastProteinLevel
Введение.
Современные масс-спектрометрические методы позволяют измерить количество белков в различных организмах. Однако эти методы имеют ограничения, такие как их сложность, стоимость и неспособность обнаружить все белки, особенно с различными физико-химическими свойствами. Более того, абсолютная количественная оценка в основном ограничивается модельными видами, что доставляет трудности исследованиям системной биологии на немодельных видах, включая многие бактерии, представляющие биотехнологический интерес.
Предсказание количества экспрессируемого белка может преодолеть эти ограничения, обеспечивая экономически эффективный, комплексный и масштабируемый подход к пониманию динамики количества белка. Это особенно важно для бактерий, где обилие белка может напрямую влиять на эффективность процессов биосинтеза. Более того, интеграция предсказания содержания белка с метаболическими моделями масштаба генома может повысить точность моделирования, позволяя более точно прогнозировать бактериальные фенотипы в различных условиях. Это может помочь в разработке генетических модификаций для оптимизации процессов биосинтеза.
Таким образом, прогнозирование изобилия белка имеет большие перспективы для развития биотехнологии и биосинтеза у бактерий. Использование современных подходов машинного обучения может улучшить понимание количественной динамики белков, оптимизировать процессы биосинтеза и ускорить разработку новых биотехнологических приложений.
Тем более, существующие методы глубокого обучения, такие как нейросетевые модели обработки естественного языка с архитектурой Transformer, обученные на больших массивах данных, позволяют анализировать биологические последовательности без ручного извлечения признаков последовательности, что значительно повышает точность предсказаний моделей машинного обучения.
В данной работе мы разработали метод предсказаний количества белка, названный yeastProtPred, включающий в себя трансформеры для получения векторных представлений последовательностей гена, а также последовательностей кодируемого белка.
Метод.
Для получения векторных представлений последовательностей генов использовался предобученный BERT-образный трансформер GENA-LM с 12 слоями и со 110 миллионами параметров, обученный на полном геноме человека Human T2T v2, расширенной геномными вариациями из gnomAD, и на полногеномных данных разных организмов из Ensembl 108.
Векторные представления последовательностей белков получались с помощью предобученного трансформера ESM-2 с 33 слоями и с 650 миллионами параметров, обученного на последовательностях белков из UniProt.
В качестве регрессионной модели использовалась библиотека градиентного бустинга CatBoost.
Результат.
На вход всех моделей подаются генетические последовательности либо белковые последовательностей или вместе. После из последних слоев трансформеров (в GENA-LM c 12 слоя, в ESM-2 с 33 слоя) извлекаются векторные представления входных последовательностей при этом веса трансформеров при обучении регрессионной модели находятся в замороженном состоянии. Полученные представления для каждого токена усреднялись по всем токенам, таким образом, получая векторные представлений одинаковой длины для всех последовательностей. Затем эти представления объединяются специальным образом и подаются на вход регрессионной модели CatBoost, которая предсказывает уровень экспрессии.
При этом, для каждой модели обучение проводилось только для регрессионной модели CatBoost при стандартных параметрах обучения, при этом выборка данных случайным образом делилась на обучающую и тестовую выборки: обучающая выборка содержала 90% всех данных, а тестовая - 10%. В качестве метрики обучения использовалась функции ошибки RMSE (корень среднеквадратичной ошибки). Далее для всех моделей проводилась кросс-валидация.
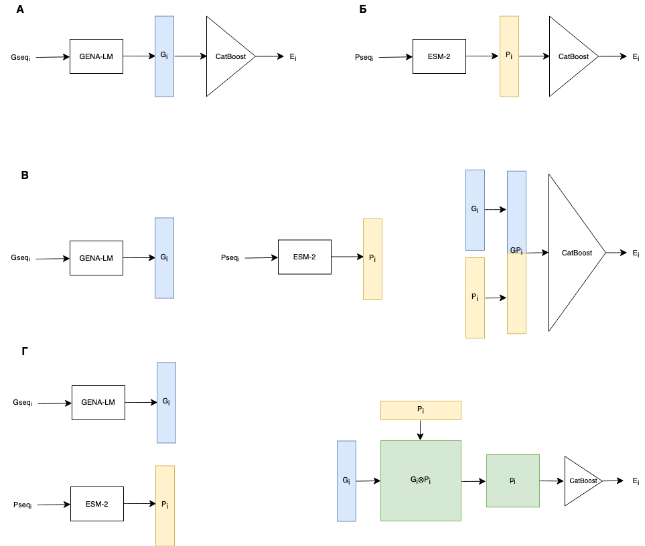
Рис.1. Схема моделей предсказания уровня экспрессии. A - GeneOnly, Б - ProtOnly, В - GeneProt, Г- GeneProtOuter16 и GeneProtOuter32, Gseqi и Pseqi - генетические и белковые последовательности i-ого гена соответственно, Gi и Pi - векторные представления генетических и белковых последовательностей, GPi - конкатенированный вектор последовательностей, Gi ⊗ Pi - внешнее произведение представлений, pi - матрица внешнего произведенеия меньше размерности, Ei - уровень экспрессии i-ого гена.
Первая модель GeneOnly (Рис.1.А) включала в себя только генетический трансформер GENA-LM и регрессионную модель CatBoost, а на вход подавались только генетические участки. Вторая модель ProtOnly (Рис.1.Б) аналогична первому, кроме того что включала в себе только белковый трансформер ESM-2 вместо генетического трансформера. В третьей модели GeneProt (Рис. 1.В) отдельно получались представления генетической и белковой последовательностей в соответствующих трансформерах, затем данные представления конкатенировались с друг другом. В четвертой и в пятой моделях GeneProtOuter16, GeneProtOuter32 (Рис.1.Г) вычислялось внешнее произведение векторных представлений, затем для каждой квадратной подматрицы заданного размера (в GeneProtOuter16 - 16x16, а в GeneProtOuter32 - 32x32) из полученной матрицы вычислялось либо среднее либо максимум. Значение максимума либо среднего затем использовалось для получения одномерного вектора меньшей длины, который подавался на вход регрессионной модели. Это было сделано из-за большого размера матрицы из результата произведения, малого размера выборки данных и недостатка вычислительных ресурсов.