Конвейер MultiDeCA для мульти-модельного de novo поиска мотивов и их комбинаторного анализа на основе данных ChIP-seq
ВВЕДЕНИЕ. Регуляция транскрипции генов эукариот осуществляется транскрипционными факторами (ТФ), которые распознают и связываются с определёнными последовательностями ДНК – сайтами связывания транскрипционных факторов (ССТФ). Поиск и анализ ССТФ имеет ключевое значение для понимания механизмов регуляции генов, так как именно через взаимодействие с сайтами связывания ТФ могут активировать или репрессировать экспрессию генов. Технология иммунопреципитации хроматина с последующим массовым секвенированием (ChIP-seq) кардинально расширила возможности картирования ССТФ в масштабах генома. ChIP-seq эксперименты генерируют тысячи геномных локусов, соответствующих районам связывания исследуемого транскрипционного фактора, что требует специализированных биоинформатических подходов для комплексного анализа полученных данных. Анализ данных ChIP-seq включает несколько этапов. Сначала выполняется картирование секвенированных прочтений на референсный геном (read mapping). Затем проводится поиск пиков (peak calling) – идентификация геномных локусов, где ТФ был связан с ДНК на основе значимого обогащения прочтений по сравнению с контролем. Третий этап – de novo поиск мотивов, то есть выявление обогащенных нуклеотидных паттернов (мотивов), характерных для комплексов ТФ–ДНК в найденных пиках. Наконец, выполняется анализ полученных мотивов ССТФ, включая их сопоставление с известными мотивами, поиск композиционных элементов (КЭ) — пар пространственно сближенных сайтов целевого и партнёрского ТФ, и биологическую интерпретацию. Учитывая колоссальный объём накопленных экспериментальных данных ChIP-seq, критическую актуальность, приобретает разработка конвейерных методов анализа, которые бы охватывали все этапы анализа данных. Для решения этой проблемы разработан новый конвейер MultiDeCA (Multi-model De novo discovery of motifs and their Combinatorial Analysis) который включает, как модули для первичной подготовки данных, так и модули для поиска мотивов разными моделями, повышая полноту детекции сайтов целевого фактора, и модули для поиска КЭ и потенциальных партнёрских ТФ. ИСПОЛЬЗУЕМЫЕ МЕТОДЫ И АЛГОРИТМЫ. (1). Картирование коротких прочтений ChIP-seq на референсный геном осуществляется с использованием высокопроизводительного алгоритма Bowtie2, обеспечивающего эффективное индексирование генома и точное выравнивание прочтений с учётом возможных нуклеотидных замен. (2). Определение геномных локусов связывания ТФ выполняется с помощью алгоритма MACS2. Метод основан на моделировании сдвига размера фрагментов ДНК и использовании распределения Пуассона для оценки статистической значимости обогащения сигнала в экспериментальном образце относительно контроля. (3). Чтобы нивелировать систематические ошибки, связанные с нуклеотидным контекстом, и минимизировать долю ложноположительных предсказаний при проведении de novo поиска мотивов необходима корректная негативная выборка. Для этой цели использовали инструмент AntiNoise, который в отличие от генерации синтетических последовательностей, извлекает из референсного генома участки, строго соответствующие позитивным пикам по длине и GC-составу. (4). Систематизация мотивов осуществляется инструментом BSMotif, который производит кластеризацию известных матриц из баз данных (HOCOMOCO, JASPAR) на основе метрик их сходства. Уникальной особенностью метода является интеграция результатов кластеризации с иерархической классификацией TFClass, что позволяет связывать вариабельность мотивов со структурными особенностями семейств и подсемейств ДНК-связывающих доменов транскрипционных факторов. (5). Идентификация мотивов производится посредством интегрированного применения четырёх моделей различной алгоритмической сложности (PWM, BaMM, InMoDe и SiteGA), что обеспечивает выявление широкого спектра структурных вариантов сайтов связывания. (6). Для объективной оценки предсказательной способности каждой модели применяется процедура двукратной перекрестной проверки. На данном этапе производится оптимизация параметров моделей и расчет метрики качества классификации: частичной площади под ROC-кривой (auROC). (7). Для обеспечения сопоставимости результатов, полученных с помощью методологически различных моделей, используется единый статистический подход к определению порогов функций распознавания. Пороги устанавливаются на основе фиксированного уровня ожидаемой частоты ложноположительных срабатываний (FPR), рассчитываемой на полногеномной выборке промоторных последовательностей. (8). Выявление потенциальных функциональных взаимодействий между транскрипционными факторами осуществляется инструментом MCOT. Алгоритм анализирует статистическую значимость совместной встречаемости пар мотивов в пределах одних и тех же пиков, идентифицируя КЭ, что свидетельствует о формировании мультибелковых комплексов. (9). Инструмент MetArea применяется для выявления взаимоисключающей встречаемости мотивов, когда присутствие сайта связывания одного фактора статистически значимо ассоциировано с отсутствием сайта другого. Такой анализ позволяет идентифицировать конкурирующие регуляторные модули или случаи, когда один транскрипционный фактор способен распознавать принципиально различные структурные классы мотивов в разных геномных контекстах, либо два разных ТФ являются антагонистами по отношению к друг-другу и чаще находятся в разных пиках. ФУНКЦИОНАЛ И НАЗНАЧЕНИЕ ИПМ. ИПМ MultiDeCA предназначен для комплексного анализа ChIP-seq данных с целью выявления мотивов ССТФ и идентификации регуляторных механизмов контроля транскрипции генов. Конвейер решает следующие задачи: 1. Предварительная обработка сырых данных ChIP-seq и извлечение нуклеотидных последовательностей геномных локусов. 2. De novo поиск мотивов ССТФ с использованием нескольких алгоритмически разных моделей для выявления структурного разнообразия сайтов связывания. 3. Аннотация обнаруженных мотивов путём сравнения с известными мотивами из баз данных HOCOMOCO и JASPAR с помощью TomTom. 4. Идентификация партнёрских транскрипционных факторов через анализ совместной и взаимоисключающей встречаемости мотивов. Функциональная интерпретация результатов через анализ терминов генной онтологии. Применение MultiDeCA позволяет существенно расширить набор предсказанных сайтов связывания по сравнению с традиционными подходами, основанными исключительно на модели PWM, и обеспечивает более полное понимание регуляторных механизмов. БЛОК-СХЕМА ИПМ. Общая архитектура ИПМ MultiDeCA представлена на рисунке Ж.29. Конвейер состоит из трёх основных функциональных блоков: (1) подготовки данных ChIP-seq, (2) подготовки общих данных и (3) анализа данных ChIP-seq.
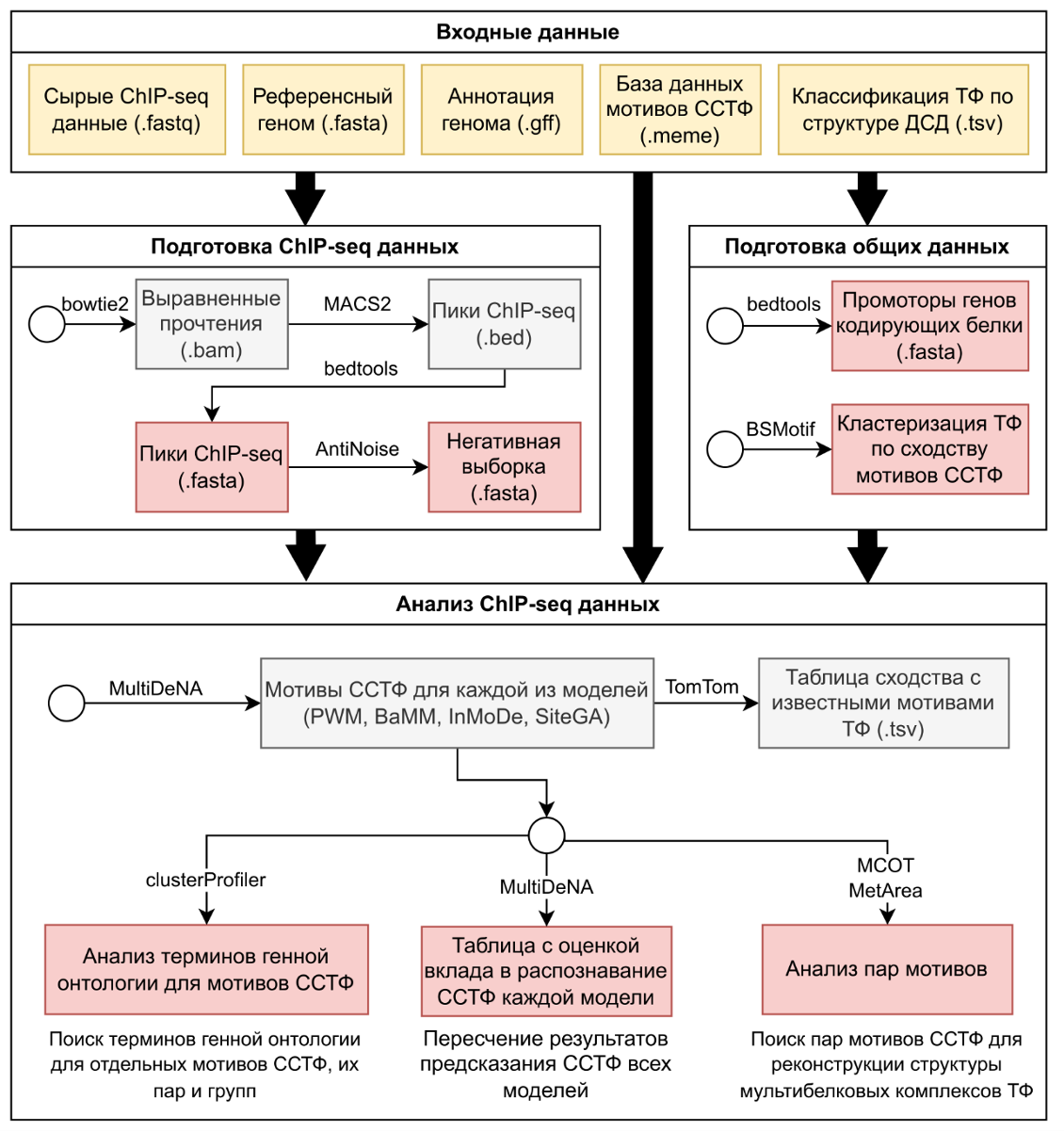
Рисунок Ж.29 – Схема конвейера MultiDeCA. Цветом обозначены виды данных: желтый – входные данные от пользователя; серый – промежуточные данные, формируемые в процессе работы конвейера; красный – выходные данные или результаты отдельных блоков. Толстыми/тонкими стрелками показаны переход данных между разными блоками/обработка некоторым инструментом (анализ или преобразование)
Блок подготовки данных ChIP-seq осуществляет предварительную обработку сырых данных и включает следующие этапы. На первом этапе производится картирование прочтений из формата FASTQ на референсный геном в формате FASTA с использованием инструмента Bowtie2. Последующее определение пиков выполняется инструментом MACS2, который идентифицирует области значимого обогащения прочтений. Из идентифицированных пиков ChIP-seq извлекаются нуклеотидные последовательности с помощью инструмента bedtools. Критически важным этапом является подготовка выборки негативных последовательностей ДНК с использованием инструмента AntiNoise. В отличие от синтетических подходов, AntiNoise извлекает геномные последовательности, точно соответствующие исходным пикам по длине и GC-составу, что обеспечивает сохранение геном-специфических особенностей и снижение доли ложноположительных результатов. Блок подготовки общих данных обеспечивает создание структурированной базы данных для аннотации результатов. Из референсного генома извлекаются последовательности промоторных районов генов (обычно от 500 до 5000 пар нуклеотидов выше сайта старта транскрипции) для последующей оценки частоты встречаемости мотивов и функциональной аннотации. На основе базы данных TFClass и используемой коллекции мотивов с помощью инструмента BSMotif создаётся иерархическая система классификации ТФ, учитывающая как структуру ДНК-связывающих доменов, так и сходство мотивов СС ТФ. Формируется полный набор ветвей транскрипционных факторов, где каждая ветвь может включать один класс, одно или несколько семейств класса, одно или несколько подсемейств, либо один или несколько транскрипционных факторов с учётом всех более низких уровней иерархии, имеющих схожие мотивы. Блок анализа данных реализует интегрированный подход к поиску и анализу мотивов СС ТФ. Инструмент MultiDeNA осуществляет de novo поиск мотивов с использованием четырёх различных моделей: PWM (реализованной в STREME), BaMM, InMoDe и SiteGA. Для каждой модели применяется процедура перекрестной проверки по схеме двукратного разделения выборки пиков на обучающую и контрольную, что позволяет подобрать оптимальные параметры модели мотива и оценить точность распознавания по метрикам auROC и auPRC. Для всех моделей единообразно определяются пороги функций распознавания исходя из заданного уровня ожидаемой частоты встречаемости мотива в полногеномной выборке промоторов, что обеспечивает сопоставимость результатов для методологически различных моделей. Обнаруженные de novo мотивы сравниваются с мотивами сайтов связывания известных транскрипционных факторов из баз данных HOCOMOCO и JASPAR с использованием инструмента TomTom. С использованием обученных мотивов производится распознавание СС ТФ в последовательностях ChIP-seq пиков. Каждое предсказание характеризуется позицией в последовательности, цепью ДНК и значением функции распознавания. Все пики классифицируются на основе наличия предсказаний: пики с предсказаниями только одной модели, пики с перекрывающимися предсказаниями нескольких моделей и пики без предсказаний. Такая классификация позволяет оценить вклад каждой модели в общее распознавание сайтов связывания целевого транскрипционного фактора. Определяется набор генов, промоторы которых содержат предсказанные ССТФ, и проводится анализ обогащения терминов Онтологии генов с помощью инструмента clusterProfiler. Инструменты MCOT и MetArea применяются для анализа пар мотивов СС ТФ. MCOT выявляет совместную встречаемость двух обогащённых мотивов в одних пиках, что отражает их присутствие в составе мультибелкового комплекса и может указывать на синергию или антагонизм транскрипционных факторов. MetArea выявляет взаимоисключающую встречаемость, при которой два обогащённых мотива разносятся в разные пики, что может отражать способность одного транскрипционного фактора связываться с различными мотивами или замену транскрипционных факторов в составе регуляторного комплекса. Входные данные для MultiDeCA: (1) Сырые данные ChIP-seq в формате FASTQ, содержащие прочтения секвенирования. (2) Референсный геном в формате FASTA для картирования прочтений и извлечения последовательностей. (3) Аннотация референсного генома в формате GFF с полногеномной разметкой генов и транскриптов. (4) База данных мотивов ССТФ в формате MEME из коллекций HOCOMOCO (человек и мышь) или JASPAR (основные таксоны эукариот). (5) Иерархическая классификация ТФ по структуре ДНК-связывающих доменов из базы TFClass в формате TSV. Выходные данные MultiDeCA: (1) Файл BED с координатами пиков ChIP-seq после обработки MACS2. (2) Файлы FASTA с нуклеотидными последовательностями пиков ChIP-seq (позитивная выборка) и негативных последовательностей (3) Мотивы ССТФ для каждой модели (PWM, BaMM, InMoDe, SiteGA) с показателями точности распознавания (auROC, auPRC) (4) Файлы BED с координатами предсказанных ССТФ для каждой модели мотива. (5) Таблица классификации пиков с указанием наличия предсказаний ССТФ различными моделями мотивов. (6) Таблицы обогащения терминов генной онтологии для генов с предсказанными ССТФ по каждой модели. (7) Таблицы значимых композиционных элементов (КЭ), выявленных инструментами MCOT и MetArea, с указанием партнёрских ТФ, взаимного расположения и ориентации сайтов. Параметры запуска задач Конвейер MultiDeCA не имеет единой интегрированной командной строки и представляет собой набор специализированных инструментов, каждый из которых запускается независимо. Для выполнения полного цикла анализа ChIP-seq данных пользователь применяет инструменты, согласно схеме (рисунок Ж.30). ПЕРЕЧЕНЬ ПРОГРАММНОГО ОБЕСПЕЧЕНИЯ И БАЗ ДАННЫХ, НЕОБХОДИМЫХ ДЛЯ РАБОТЫ ИПМ. MultiDeCA представляет собой конвейер, объединяющий несколько независимых инструментов, каждый из которых имеет собственные требования к программному окружению. Следующие инструменты необходимы для проведения анализа: 1. Bowtie2 — картирование прочтений на референсный геном (C/C++) 2. MACS2 — поиск пиков ChIP-seq (Python) 3. bedtools — манипуляции с геномными интервалами (C/C++) 4. AntiNoise — подготовка негативных выборок для de novo поиска (C/C++) 5. MultiDeNA — интеграция de novo моделей и аннотация мотивов (Python) 6. STREME — de novo поиск мотивов модели PWM (C/C++) 7. BaMM — de novo поиск байесовской-марковской модели мотива (C/C++) 8. InMoDe — de novo поиск интерполированной марковской модели мотива (Java) 9. SiteGA — de novo поиск мотива с помощью генетического алгоритма и представление его в виде набора локально-позиционированных динуклеотидов (C/C++) 10. BSMotif — кластеризация мотивов (Python) 11. MCOT — анализ совместной встречаемости мотивов (C/C++) 12. MetArea — анализ взаимоисключающей встречаемости (C/C++) 13. TomTom — сравнение мотивов (C/C++) 14. clusterProfiler — анализ обогащения генной онтологии (R) 15. HOCOMOCO — мотивы сайтов связывания транскрипционных факторов человека и мыши (.meme) 16. JASPAR — мотивы для основных таксонов эукариот (.meme) 17. TFClass — иерархическая классификация транскрипционных факторов по структуре ДНК-связывающих доменов (.tsv) Установка отдельных инструментов конвейера производится согласно их индивидуальной документации. Для инструментов на Python используется менеджер пакетов pip или conda. Для инструментов, реализованных на языке R, установка осуществляется посредством стандартных механизмов распространения пакетов, включая репозитории CRAN и Bioconductor, а также встроенные средства управления пакетами среды R. Для инструментов, разработанных на C/C++, возможны различные подходы к установке: использование предварительно собранных бинарных пакетов (например, из репозиториев Linux-дистрибутивов или каналов conda) либо сборка из исходного кода, размещённого в специализированных репозиториях, с последующей установкой в соответствии с предоставленной разработчиками документацией. ИСХОДНЫЕ КОДЫ ИПМ. Исходные коды инструментов, составляющих конвейер MultiDeCA, доступны в открытом доступе (см. зависимости). Основные инструменты MultiDeNA, BSMotif, MCOT, MetArea и AntiNoise разработаны в Институте цитологии и генетики СО РАН и доступны через репозитории разработчиков. Коды вспомогательных инструментов (Bowtie2, MACS2, bedtools, STREME, BaMM, InMoDe, TomTom, clusterProfiler) доступны через официальные репозитории. ТЕСТОВЫЕ ПРИМЕРЫ. Демонстрационные наборы данных поставляются вместе с отдельными программами, входящими в конвейер, и предназначены для проверки корректности установки и базовой работоспособности каждого инструмента. Набор данных для запуска всего конвейера за исключением этапа конвейера можно взять в репозитории MultiDeNA (в директории example), оно включает подмножество заранее подготовленных пиков ChIP-seq и соответствующий референсный геном. ДЕМОНСТРАЦИОННЫЕ ПРИМЕРЫ РЕШЕНИЯ СОДЕРЖАТЕЛЬНЫХ ЗАДАЧ. Для демонстрации возможностей конвейера MultiDeCA был проведён анализ ChIP-seq данных по транскрипционному фактору JunD. Данные эксперимента получены на макрофагах костного мозга мыши, обработанных 100 нг/мл липидом A в течение 30 минут (GEO ID GSM2663855, GTRD ID PEAKS040980). Исходные данные были представлены в виде 107026 пиков, идентифицированных с помощью MACS2. Для de novo поиска мотивов были отобраны 1000 наиболее значимых пиков (с наибольшими значениями –log10(p-value) по MACS2). Такой отбор сделан с целью сосредоточиться на высококачественных сайтах связывания и снизить вычислительную нагрузку. Подготовка данных. С помощью bedtools извлечены нуклеотидные последовательности пиков из полного генома. Инструмент AntiNoise подготовил негативную выборку последовательностей, соответствующих позитивным по длине и GC-составу. De novo поиск мотивов. Инструмент MultiDeNA осуществил подбор параметров моделей мотива PWM, BaMM и SiteGA с помощью перекрестной проверки и выполнил de novo поиск мотивов этих моделей. Все три обнаруженных мотива продемонстрировали значимое сходство (TomTom p-value < 0.0001) с мотивами сайтов связывания, относящимися к той же ветви транскрипционных факторов, что и целевой фактор JunD согласно кластеризации BSMotif. Анализ структуры мотивов показал, что для всех трёх моделей совпадают первые три консервативных нуклеотида TGA (рисунок Ж.30А).
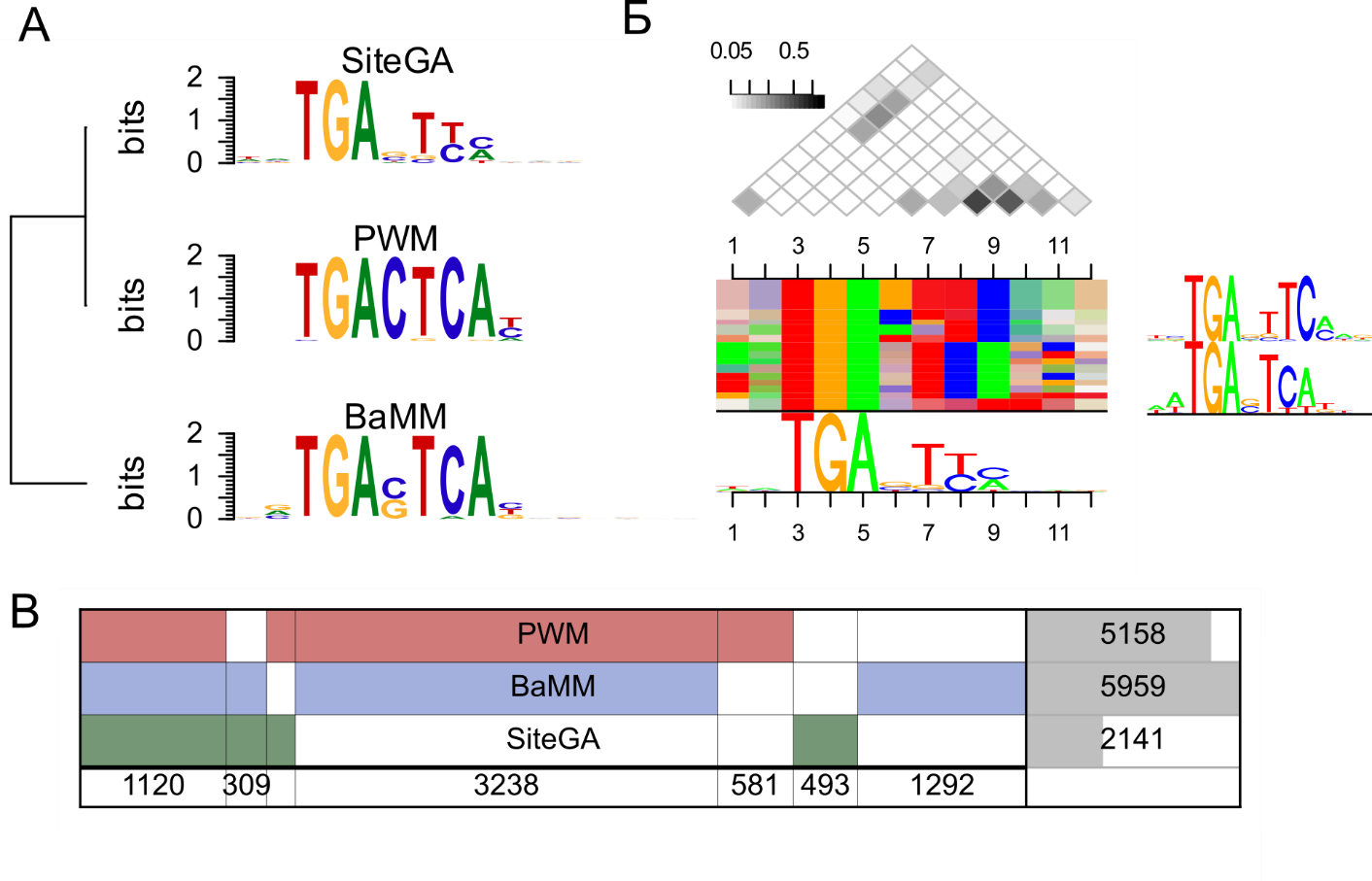
Рисунок Ж.30 – Результаты работы конвейера MultiDeCA. (А) Лого-диаграммы для моделей мотива PWM, BaMM и SiteGA. (Б) DepLogo (Grau et al., 2019) включает лого-диаграммы (справа и внизу), цветные полосы в центре и треугольную матрицу сверху. Цветные полосы отражают частоты нуклеотидов в позициях мотива. Треугольная матрица отражает зависимости разных позициями с помощью метрики взаимной информации, отмеченной оттенками серого цвета. В правой части - две лого-диаграммы, отражающие два структурных типа ССТФ, предсказанных моделью SiteGA, по длине спейсере в один или две пары оснований. (В) Диаграмма SuperVenn, отображающая пересечения множеств пиков с предсказанными ССТФ JunD моделями PWM (красный), BaMM (синий) и SiteGA (зеленый). Каждая строка соответствует одной модели, а каждый столбец — уникальной комбинации (пересечению) этих моделей. Наличие цветного блока в столбце означает, что в данных пиках были предсказаны ССТФ соответствующей моделью (например, синий цвет маркирует участие модели BaMM в пересечении). Числа справа на сером фоне показывают общее количество пиков с предсказанными ССТФ каждой моделью. Числа внизу указывают количество пиков в каждом конкретном пересечении (столбце); значения приведены только для пересечений размером более 300 пиков Мотив, выявленный моделью SiteGA продемонстрировал пониженный уровень консервативности на правом фланге, что при дополнительном анализе с помощью DepLogo объясняется предсказанием ССТФ с вариабельным спейсером длиной 1 или 2 пары нуклеотидов между левым TGA и правым TCA флангами (рисунок Ж.30Б). Эта особенность отражает биологическую реальность связывания транскрипционного фактора JunD, относящегося к классу Basic leucine zipper factors (bZIP), исключительно в форме димера с допустимой вариацией длины спейсера.
СПИСОК ПУБЛИКАЦИЙ
- Tsukanov A.V., Levitsky V.G., Merkulova T.I. Application of alternative de novo motif recognition models for analysis of structural heterogeneity of transcription factor binding sites: a case study of FOXA2 binding sites // Vavilov Journal of Genetics and Breeding. 2021. Vol. 25(1). P. 7–17.
- Tsukanov A.V., Mironova V.V., Levitsky V.G. Motif models proposing independent and interdependent impacts of nucleotides are related to high and low affinity transcription factor binding sites in Arabidopsis // Frontiers in Plant Science. 2022. Vol. 13. Art. 938545.
- Raditsa V.V., Tsukanov A.V., Bogomolov A.G., Levitsky V.G. Genomic background sequences systematically outperform synthetic ones in de novo motif discovery for ChIP-seq data // NAR Genomics and Bioinformatics. 2024. Vol. 6(3). Art. lqae090.
- Levitsky V.G., Zemlyanskaya E.V., Oshchepkov D.Yu. et al. A single ChIP-seq dataset is sufficient for comprehensive analysis of motifs co-occurrence with MCOT package // Nucleic Acids Research. 2019. Vol. 47(21). Art. e139.
- Levitsky V.G., Oshchepkov D.Yu., Zemlyanskaya E.V., Merkulova T.I. Asymmetric conservation within pairs of co-occurred motifs mediates weak direct binding of transcription factors in ChIP-Seq data // International Journal of Molecular Sciences. 2020. Vol. 21(17). Art. 6023.
- Levitsky V.G., Tsukanov A.V., Merkulova T.I. MetArea: a software package for analysis of the mutually exclusive occurrence in pairs of motifs of transcription factor binding sites based on ChIP-seq data // Vavilov Journal of Genetics and Breeding. 2024. Vol. 28(8). P. 822–833.
- Levitsky V.G., Raditsa V.V., Tsukanov A.V. et al. Asymmetry of motif conservation within their homotypic pairs distinguishes DNA-binding domains of target transcription factors in ChIP-Seq data // International Journal of Molecular Sciences. 2025. Vol. 26(1). Art. 386.
- Levitsky V.G., Vatolina T.Yu., Raditsa V.V. Linking hierarchical classification of transcription factors by the structure of their DNA-binding domains to the variability of their binding site motifs // Vavilov Journal of Genetics and Breeding. 2025. Vol. 29(7). P. 925–939.