MetArea для выявления
ЗАДАЧИ МОДУЛЯ. Компьютерный анализ особенностей прямого и непрямого (через белок-посредник) связывания транскрипционных факторов с геномной ДНК (с учётом открытого и закрытого хроматина); Разработка конвейера, интегрирующего программное обеспечение, созданное для анализа ChIP-seq данных; поиск мотивов сайтов связывания транскрипционных факторов на основе разработанного конвейера ПРЕДНАЗНАЧЕНИЕ ПРОГРАММНОГО МОДУЛЯ «METAREA». Программный модуль MetArea предназначен для выявления пар мотивов сайтов связывания транскрипционных факторов (ССТФ) со взаимоисключающей встречаемостью. Когда по заданному набору данных массового секвенирования ChIP-seq определены обогащённые мотивы, предположительно представляющие ССТФ, на механизмы действия транскрипционных факторов (ТФ) могут указать статистические закономерности встреч мотивов в парах. Однако обогащённые мотивы, выявляемые с помощью de novo поиска мотивов, не обязательно принадлежат только ССТФ для исследуемого в эксперименте (целевого) ТФ. ТФ in vivo могут связываться с ДНК разными способами: • напрямую (в ДНК есть сайт связывания целевого ТФ); • при помощи другого «партнёрского» ТФ (в ДНК есть сайты связывания целевого и партнёрского ТФ встречаются совместно (рядом)) с некоторым спейсером или перекрываются); • не напрямую (в ДНК есть сайт связывания партнёрского ТФ и нет сайта связывания целевого ТФ). Ранее были определены понятия синергии и антагонизма ССТФ в составе композиционного элемента (КЭ), устойчивого сочетания для пары ССТФ (Kel et al., 1995). Синергия для двух конкретных ССТФ в регуляторном районе гена означает, что результат действия пары ТФ значительно превосходит результат действия каждого из них в отдельности. При антагонизме ТФ, наоборот, друг другу мешают. Например, один из двух ТФ активатор, а другой репрессор, так что один из них вытесняет другого. Однако, понятия синергия и антагонизм относятся в данном случае к устойчивой паре двух встречаемых в ДНК сайтов, и эти два случая невозможно различить по частотам совместных встреч в паре мотивов, представляющих ССТФ. В случае данных ChIP-seq бионформатический анализ имеет дело с набором сотен или даже тысяч геномных локусов, в которых в целом наблюдается как прямое, так и непрямое связывание целевого ТФ. При переходе от отдельного рассмотрения частот двух мотивов ССТФ в наборе пиков ChIP-seq к закономерностям связи между этими двумя мотивами целесообразно рассматривать две возможности: • два мотива чаще, чем ожидается по случайным причинам, встречаются вместе в одних пиках и реже по отдельности в разных пиках, • два мотива встречаются чаще в разных пиках и реже в одних пиках. Поэтому для пары мотивов ССТФ предложены термины совместной и взаимоисключающей встречаемости (Levitsky et al., 2024, рисунок Ж.26).
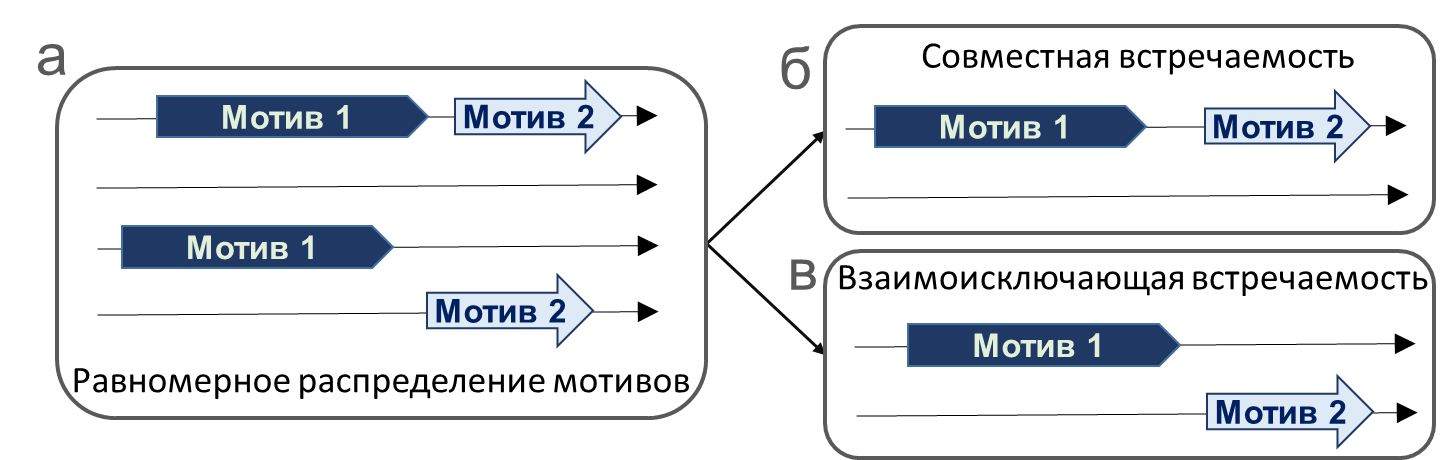
Рисунок Ж.26 – Определение терминов совместной и взаимоисключающей встречаемости мотивов ССТФ. Пусть частота встреч каждого из двух мотивов в пике 50%. (а) Два мотива появляются в пиках независимо друг от друга, есть четыре равновероятные варианта картирования мотивов в пиках. (б) Совместная встречаемость в пике есть только сайты обеих мотивов или нет ни одного из двух. (в) Взаимоисключающая встречаемость в пике есть только сайт одного мотив из двух мотивов. Стрелки от панели (а) к панелям (б) и (в) означают, что четыре варианта панели (а) точно разделяются на две группы по два варианта на панелях (б) и (в) Программный модуль MetArea производит расчёты оценок точности распознавания мотива pAUPRC (частичная площадь под кривой PR) для каждого из двух входных одиночных мотивов, а также для их сочетания, «объединённого мотива» (Levitsky et al., 2024). Алгоритм MetArea рассматривает два входных «одиночных» мотива, а также некий «объединённый» мотив, означающий появление любого их двух одиночных. Для распознавания объединённого мотива в последовательности ДНК достаточно распознавания в ней хотя бы одного из двух одиночных мотивов при заданном пороге ожидаемой частоты мотива. Для оценки точности модели распознавания мотива мы разработали и применили меру точности «Частичная площадь под кривой PR (Precision – Recall)», для расчёта которой нужно отслеживать число распознанных последовательностей позитивной и негативных выборок. Кривая PR это зависимость меры Precision, при равных объёмах выборок NF и NB Precision = TP/(TP + FP) (отношения числа предсказанных последовательностей в позитивной выборке к числу предсказанных последовательностей в позитивной и негативной выборках) от меры Recall = TP/NF (отношения числа предсказанных последовательностей позитивной выборки к общему числу последовательностей этой выборки, то есть объёму выборки). При неравных объёмах выборок расчёт величины Precision происходит следующим образом:
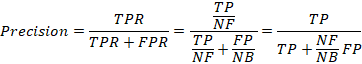
Схема расчёта величины pAUPRC частичной площади под кривой PR представлена на рисунке Ж.27.
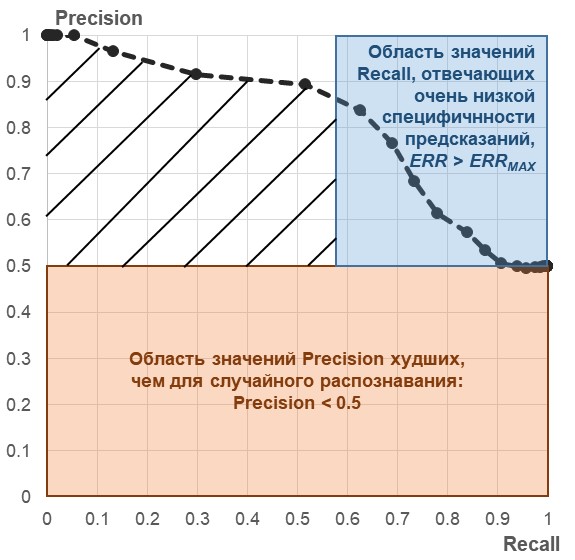
Рисунок Ж.27 – Схема расчёта частичной площади под кривой PR. Ось X – мера Recall (вероятность предсказания последовательности позитивной выборки, Recall = TPR = TP/NF). Ось Y – мера Precision, отношение вероятности предсказания последовательности позитивной выборки к сумме вероятностей предсказания последовательностей позитивной и негативной выборок, Precision = TPR/(TPR+FPR). Розовая область отмечает значения Precision < 0.5, соответствующие предсказаниям худшим, чем предсказания случайной модели, равновероятно распознающей последовательности позитивной и негативной выборок. Precision > 0.5 / Precision < 0.5 области отбора в строну позитивной / негативной выборок. Голубая область показывает область предсказанных последовательностей позитивной выборки с очень низкой специфичностью, соответствующих ожидаемой частоте мотива большей порога, ERR > ERRMAX. Для генерации данных примера негативной выборки взято нормальное распределение со средним и стандартным отклонением (N, N) = (5, 2.5), а позитивная выборка это смесь 50% на 50% нормальных распределений (P1, P1) = (10, 1) и (P2, P2) = (5.5, 4). Эти распределения моделируют сайты, проходящие и не проходящие порог ERRMAX ожидаемой частоты мотива. Штриховка обозначает область, по которой определяется метрика частичной площади под кривой pAUPRC
Критерий предсказания функциональной связи мотивов отражает повышение оценки точности объединённого мотива по сравнению с оценками точности одиночных мотивов, этот критерий является количественной оценкой взаимоисключающей встречаемости в парах мотивов. Для пары мотивов A и B критерий требует более высокого значения оценки точности pAUPRC(A&B) объединённого мотива A&B по сравнению со значениями оценок точностей обоих одиночных мотивов, pAUPRC(A) и pAUPRC(B). Рассчитанное таким образом Отношение Площадей Под Кривыми, ОППК (Ratio of Areas Under Curves, RAUC) должно быть больше единицы:
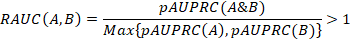
Общая схема работы алгоритма MetArea представлена на рисунке Ж.28.
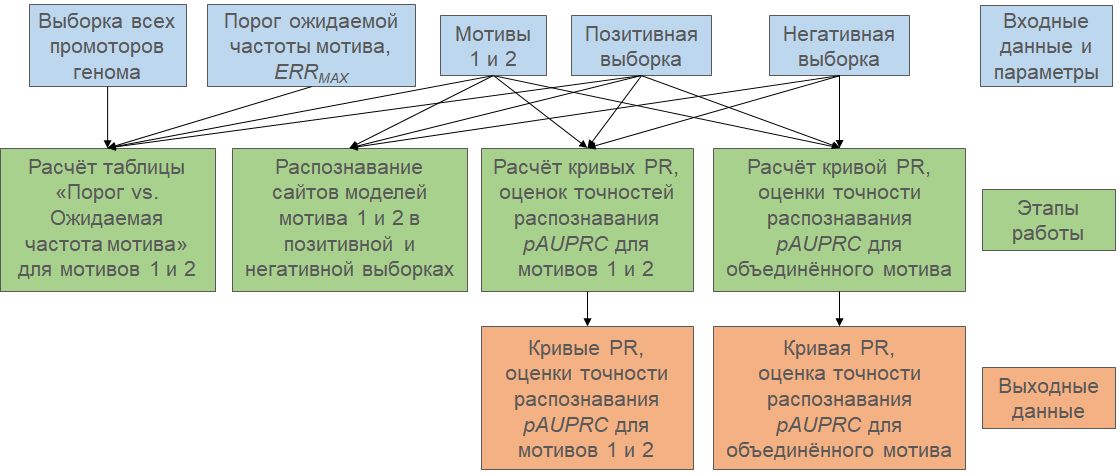
Рисунок Ж.28 – Схема работы модуля MetArea
ОПИСАНИЕ ФУНКЦИОНАЛА ПРОГРАММНОГО МОДУЛЯ, ПАРАМЕТРЫ, ВХОДНЫЕ И ВЫХОДНЫЕ ДАННЫЕ. Входными данными и параметрами программного модуля MetArea являются: • Два мотива ССТФ, возможны варианта моделей этих мотивов: (1) сочетание двух мотивов традиционной модели ПВМ (позиционной весовой матрицы, Wasserman, Sandelin, 2004), заданными двумя матрицами частот нуклеотидов (МЧН), или (2) сочетание мотива модели ПВМ, заданной МЧН и альтернативной модели мотива SiteGA, заданной своей весовой матрицей, см. программный модуль SiteGA, https://github.com/parthian-sterlet/sitega (Tsukanov et al., 2022); (3) сочетание двух мотивов модели SiteGA. • Позитивная выборка в формате FASTA (набор пиков ChIP-seq, NF последовательностей, Number of Foreground sequences). • Негативная выборка в формате FASTA (NB последовательностей, Number of Background sequences), её рекомендуется приготовить предварительно по позитивной выборке и полному геному соответствующего вида организма с помощью программного модуля AntiNoise (Raditsa et al., 2024), https://github.com/parthian-sterlet/antinoise. Для каждой последовательности позитивной выборки по её длине и G/C-составу в полном геноме случайным образом находится несколько последовательностей негативной выборки. Параметр программного модуля AntiNoise по умолчанию NF / NB = 5. • Выборка промоторов всех белок-кодирующих генов генома, необходимая для определения порогов распознавания на основе расчёта таблиц ‘Threshold vs. ERR’ («Порог функции распознавания vs. Частота мотива в выборке всех промоторов генома») для каждого из входных мотивов. В составе программного комплекса есть выборки промоторов для основных модельных видов эукариот, см. программный модуль AntiNoise. • Порог ERRMAX максимальной ожидаемой частоты для каждого входного мотива (Expected Recognition Rate, ERR). Рекомендуются значения в диапазоне от 0.001 до 0.01, значение по умолчанию 0.002. • Таблицы ‘Threshold vs. ERR’ для каждого входного мотива. Максимальная частота мотива 0.01 означает, что специфичность CCТФ соответствует одному сайту на сто пар оснований. Мы ранее использовали таблицы ‘Threshold vs. ERR’ для унификации порогов распознавания разных мотивов (Levitsky et al., 2019; Tsukanov et al., 2022). Каждый мотив вместе со своей таблицей ‘Threshold vs. ERR’ подаётся в файле бинарного формата, генерируемого компонентами программного модуля MetArea для расчёта ожидаемых частот мотива для моделей мотива ПВМ и SiteGA. ОБЩЕЕ ОПИСАНИЕ ВХОДНЫХ ДАННЫХ. На вход программного модуля подаются: • путь к исполняемым файлам; • путь к выборкам промоторов всех генов генома кодирующих белок (для видов человек, мышь, арабидопсис, дрозофила); • два или более входных мотива заданных моделей, либо для одного, обеих или нескольких мотивов путь к папке с библиотекой мотивов, включающей для каждого мотива его МЧН и таблицу ERR; • путь к папке с выходными данными. Общие параметры запуска Порог ERRMAX максимальной ожидаемой частоты для каждого входного мотива (Expected Recognition Rate, ERR). С помощью этого порога, а также выборки промоторов всех генов генома кодирующих белки для каждого входного мотива рассчитывается таблица ‘Threshold vs. ERR’. Эта таблица включает две колонки: величина порога функции распознавания и доля проверенных позиций в выборке, содержащих предсказанные сайты. Описание общих выходных данных • Текстовый файл с кривыми PR для каждого из входных мотивов, а также их объединённого мотива. • Текстовый файл со значениями оценок точности распознавания pAUPRC для каждого из входных мотивов, а также их объединённого мотива, значением отношения площадей под кривыми (см. ниже), оценкой сходства мотивов (только для пар мотивов модели ПВМ). АЛГОРИТМИЧЕСКАЯ РЕАЛИЗАЦИЯ, КОДЫ И АДРЕС ПРОГРАММНОГО МОДУЛЯ. Модуль реализован на языке с++. Конвейеры запуска реализованы на языке perl. Исходный код, конвейерные скрипты и примеры их запуска доступны по адресу HTTPS://GITHUB.COM/PARTHIAN-STERLET/METAREA. СПИСОК И ОПИСАНИЕ ВСПОМОГАТЕЛЬНЫХ ПРОГРАММ, ВЫПОЛНЯЮЩИХ ЭТИ ДОПОЛНИТЕЛЬНЫЕ ЭТАПЫПредобработка входных данных может включать следующие дополнительные этапы: • Расчёт таблиц ‘Threshold vs. ERR’ для одного или нескольких входных мотивов в текстовом виде и запись их в текстовом и бинарном формате (для моделей мотива ПВМ и SiteGA генерируются программой выбора порогов ПВМ из MCOT (pwm_iz_pwm_thr_dist0.cpp, Levitsky et al., 2019) и программой выбора порогов SiteGA из модуля SiteGA (sitega_thr_dist_mat.cpp, Tsukanov et al., 2022). • Создание бинарного файла для модели ПВМ из текстовых файлов матриц МЧН/ПВМ и таблицы ‘Threshold vs. ERR’, pwm_text_binary_write.cpp • Создание бинарного файла для модели SiteGA из текстового файла матрицы и таблицы ‘Threshold vs. ERR’, pwm_sga_text_binary_write.cpp Список программ для компиляции и сборки модуля • ОС Линукс • Компилятор С++ GCC • Команды загрузки, установки и компиляции модуля в ОС Linux git clone https://github.com/parthian-sterlet/MetArea cd MetArea/runchmod a+x build.sh ./build.sh ВАРИАНТЫ ПРИМЕНЕНИЯ С ПРИМЕРАМИ И ТЕСТОВЫМИ ВХОДНЫМИ ДАННЫМИ. Входными данными программного модуля MetArea могут быть мотивы ССТФ, для которых предполагается обогащение в позитивной выборке по сравнению с негативной, например, такие мотивы — это результаты de novo поиска мотивов (Bailey, 2021). Отдельные варианты применения программного модуля реализуют массовый анализ коллекций мотивов ССТФ из баз данных Hocomoco (Vorontsov et al., 2024) и JASPAR (Rauluseviciute et al., 2024). Анализ множества пар мотивов позволяет выявить пары, в которых выявляется наибольший рост оценки точности распознавания pAUPRC при объединении мотивов. Разные варианты применения рассматривают мотивы моделей ПВМ и SiteGA, эти мотивы могут быть взяты из результатов de novo поиска мотивов, или из баз данных. Программный модуль допускает следующие варианты применения: 1. Два мотива модели ПВМ; 2. Два мотива, модель ПВМ модель SiteGA; 3. Несколько мотивов модели ПВМ; Эти мотивы могут быть результатами de novo поиска. Для всех K входных мотивов проверяются все возможные {K * (K − 1)/2} пары мотивов. Вариант реализован с помощью скрипта на языке Perl, использующего вспомогательные и основные программы программного модуля. 4. Мотив модели ПВМ против всей коллекции (M) мотивов CCТФ из базы данных. Для заданного мотива проверяются все его M пар с мотивами из коллекции Hocomoco (человек, мышь) или JASPAR (растения, насекомые). 5. Лучшие мотивы из всей коллекции мотивов CCТФ из базы данных. Из всех N мотивов из коллекции базы данных Hocomoco или JASPAR отбираются M мотивов с наивысшими оценками точности pAUPRC, и проверяются все возможные пары с участием этих мотивов, их всего {M * (M − 1)/2}. Доступны несколько коллекций мотивов из баз данных мотивов ССТФ: • коллекции 1420/1142 мотивов для 942/713 ТФ человека/мыши из базы данных Hocomoco версии 12 (Vorontsov et al., 2024); • 556/151 мотивов для 555/148 растений/насекомых из базы данных JASPAR (Rauluseviciute et al., 2024). Списки аргументов командной строки для разных вариантов применения программного модуля «Два мотива модели ПВМ», исходный код pauc_forback_2motifs_only.cpp 1. входной файл FASTA, первый набор последовательностей ДНК (позитивная выборка), пики ChIP-seq 2. входной файл FASTA, второй набор последовательностей (негативная выборка), геномные последовательности, сопоставленные с пиками в содержании А/Т и длина, извлеченные с помощью программного модуля AntiNoise 3. входной бинарный файл для первого мотива (ПВМ1). Этот файл интегрирует в бинарный формат МЧН, ПВМ, длина мотива и общее количество порогов распознавания, полный список порогов и значения ERR. Бинарные файлы для моделей ПВМ генерируются программой выбора порогов ПВМ из MCOT (https://github.com/parthian-sterlet/mcot-kernel, Levitsky et al., 2019). 4. входной бинарный файл для второго мотива (ПВМ2), требования аналогичны аргументу №3. 5. вещественное число, порог ERRMAX, рекомендуются значения в диапазоне от 0.001 до 0.01, значение по умолчанию – 0.002. Значение ERRMAX (максимальная ожидаемая скорость распознавания) означает самый мягкий порог, используемый для вычисления частичной области под кривой PR. Это значение ERRMAX определяет диапазон порогов распознавания из таблицы ‘Threshold vs. ERR’. 6. выходной файл, значения pAUPRC для мотивов ПВМ1 и ПВМ2 и для объединённого мотива ПВМ1&ПВМ2, значение Отношение Площадей Под Кривыми (RAUC), значение сходства между двумя мотивами, рассчитанное согласно подходу MCOT (https://github.com/parthian-sterlet/mcot-kernel, Levitsky et al., 2019) 7. выходной файл, кривая PR для мотива ПВМ1 8. выходной файл, кривая PR для мотива ПВМ2 9. выходной файл, кривая PR для объединённого мотива ПВМ1&ПВМ2 «Два мотива, модель ПВМ модель SiteGA», исходный код pauc_forback_pwm_sga_only.cpp. Число и список аргументов совпадает с соответствующими для варианта «Два мотива модели ПВМ», незначительно отличается описание только аргументов 4, 6, 8 и 9. 4. входной бинарный файл для второго мотива (SiteGA). Этот файл интегрирует в бинарный формат матрицу модели SiteGA, включая длину мотива, общее количество порогов распознавания, полный список порогов и значений ERR. Бинарный файл для модели SiteGA генерируется программой выбора порогов из модуля SiteGA, исходный код sitega_thr_dist_mat.cpp 6. выходной файл, значения pAUPRC для мотивов ПВМ и SiteGA для объединённого мотива ПВМ&SiteGA, значение Отношение Площадей Под Кривыми (RAUC). 8. выходной файл, кривая PR для мотива SiteGA 9. выходной файл, кривая PR для объединённого мотива ПВМ&SiteGA «Несколько мотивов модели ПВМ», исходный код pauc_forback_2motif0.cpp. Аргументы №1, 2, 5 такие же, как и у варианта «Два мотива модели ПВМ». 3. входной бинарный файл для набора N мотивов модели ПВМ. Этот бинарный файл предварительно генерируется программой выбора порогов ПВМ из MCOT, исходный код pwm_iz_pwm_thr_dist0.cpp (Levitsky et al., 2019). 4. целое число, число мотивов, N 6. выходной файл, матрицы всех {N * (N - 1)/2} попарных значений (а) оценок точности pAUPRC, (б) Отношений Площадей Под Кривыми (ОППК), (в) значения сходства МЧН (оценивается с помощью компонента программного комплекса MCOT, исходный код pwm_iz_pwm_thr_dist0.cpp (Levitsky et al., 2019). 7. выходной файл pAUPRC, список значений pAUPRC для всех пар мотивов, сформированных из N мотивов 8. выходной файл log1, список значений pAUPRC для всех отдельных мотивов 9. выходной файл log2, список значений pAUPRC для всех пар объединённых мотивов, состоящих из N мотивов 10. выходной файл, кривые PR для одиночных и объединённых мотивов, например, кривая PR для пары моделей Пример для варианта «Мотив модели ПВМ против всей коллекции (M) мотивов CC ТФ из базы данных», исходный код pauc_forback_anc_lib.cpp. Аргументы №1, 2, 3, 5 такие же, как у варианта «Два мотива модели ПВМ». 4. входной файл в бинарном формате для коллекции мотивов ПВМ. Эти файлы в бинарном формате интегрируют для всех мотивов в каждой коллекции МЧН, ПВМ, длина мотива и общее количество порогов распознавания, полный список порогов и значения ERR. Бинарные файлы для моделей ПВМ генерируются программой выбора порогов ПВМ из MCOT, исходный код pwm_iz_pwm_thr_dist0.cpp 6. выходной файл, список значений pAUPRC для объединённых мотивов, удовлетворяющих критерию Отношение Площадей Под Кривыми, ОППК > 1. Также указывается семейство и класс ТФ для мотива из базы данных. 7. выходной файл, список значений pAUPRC и ОППК объединённого мотива для всех пар мотивов, значение сходства между в парах мотивов, рассчитанное согласно подходу MCOT (Levitsky et al., 2019). Также указывается семейство и класс ТФ для мотива из базы данных. Пример для варианта «Лучшие мотивы из всей коллекции мотивов CC ТФ из базы данных», исходный код pauc_forback_2motifs.cpp. Аргументы №1, 2 такие же, как у варианта «Мотив модели ПВМ против всей коллекции (M) мотивов CCТФ из базы данных». 3. входной файл в бинарном формате для коллекции мотивов ПВМ. Эти файлы в бинарном формате интегрируют для всех мотивов в каждой коллекции МЧН, ПВМ, длина мотива и общее количество порогов распознавания, полный список порогов и значения ERR. Бинарные файлы для моделей ПВМ генерируются программой выбора порогов ПВМ из MCOT, исходный код pwm_iz_pwm_thr_dist0.cpp 4. входной файл в бинарном формате, матрица парных сравнений сходства для всех M мотивов коллекции 5. целое число, число отбираемых M наиболее высоко оцененных мотивов 6. вещественное число, порог ERRMAX, рекомендуются значения в диапазоне от 0.001 до 0.01, значение по умолчанию – 0.002. Значение ERRMAX (максимальная ожидаемая скорость распознавания) означает самый мягкий порог, используемый для вычисления частичной области под кривой PR. Это значение ERRMAX определяет диапазон порогов распознавания из таблицы ‘Threshold vs. ERR’. 7. выходной файл, матрицы всех {M * (M - 1)/2} попарных значений (а) оценок точности pAUPRC, (б) Отношений Площадей Под Кривыми (ОППК), (в) значения сходства МЧН (оценивается с помощью компонента программного комплекса MCOT, исходный код pwm_iz_pwm_thr_dist0.cpp (Levitsky et al., 2019). 8. выходной файл pAUPRC, список значений pAUPRC для всех пар мотивов, сформированных из M мотивов, для которых ОППК > 1, также указываются сходство мотивов и классы/семейства ТФ 9. выходной файл log1, список значений pAUPRC для всех отдельных мотивов 10. выходной файл log2, список значений pAUPRC для всех пар объединённых мотивов, сформированных М мотивами.
СПИСОК ЦИТИРОВАННОЙ ЛИТЕРАТУРЫ
1. Amoutzias G. D., Robertson D. L., Van de Peer Y., Oliver S. G. Choose your partners: dimerization in eukaryotic transcription factors. Trends Biochem Sci. 2008; 33(5):220–229.
2. Bailey T. L. STREME: Accurate and versatile sequence motif discovery. Bioinformatics 2021;37:2834–2840.
3. Hess D. A., Strelau K. M., Karki A., Jiang M., Azevedo-Pouly A. C., Lee A. H., Deering T. G., Hoang C. Q., MacDonald R. J., Konieczny S. F. MIST1 links secretion and stress as both target and regulator of the unfolded protein response. Mol Cell Biol. 2016;36(23):2931–2944.
4. Kel O. V., Romaschenko A. G., Kel A. E., Wingender E., Kolchanov N. A. A compilation of composite regulatory elements affecting gene transcription in vertebrates. Nucleic Acids Res. 1995;23(20):4097-4103.
5. Kolmykov S., Yevshin I., Kulyashov M., Sharipov R., Kondrakhin Y., Makeev V. J., Kulakovskiy I. V., Kel A., Kolpakov F. GTRD: An integrated view of transcription regulation. Nucleic Acids Res. 2021;49(D1):D104–D111.
6. Levitsky V.G., Tsukanov A.V., Merkulova T.I. MetArea: a software package for analysis of the mutually exclusive occurrence in pairs of motifs of transcription factor binding sites based on ChIP-seq data. Vavilov J Genet Breed. 2024; 28(8), 822-833.
7. Levitsky V. G., Vatolina T. Yu., Raditsa V. V. Linking hierarchical classification of transcription factors by the structure of their DNA-binding domains to the variability of their binding site motifs. Vavilov J Genet Breed. 2025; 29(7): 925-939.
8. Levitsky V., Zemlyanskaya E., Oshchepkov D., Podkolodnaya O., Ignatieva E., Grosse I., Mironova V., Merkulova T. A single ChIP-seq dataset is sufficient for comprehensive analysis of motifs co-occurrence with MCOT package. Nucleic Acids Res. 2019;47:e139.
9. Rauluseviciute I., Riudavets-Puig R., Blanc-Mathieu R., Castro-Mondragon J. A., Ferenc K., Kumar V., Lemma R. B., Lucas J., Chèneby J., Baranasic D., et al. JASPAR 2024: 20th anniversary of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2024;52(D1):D174-D182.
10. Raditsa V. V., Tsukanov A. V., Bogomolov A. G., Levitsky V. G. Genomic background sequences systematically outperform synthetic ones in de novo motif discovery for ChIP-seq data. NAR Genom Bioinform. 2024;6(3):lqae090.
11. Tsukanov, A.V., Mironova, V.V., Levitsky, V.G. (2022) Motif models proposing independent and interdependent impacts of nucleotides are related to high and low affinity transcription factor binding sites in Arabidopsis. Front Plant Sci. 13, 938545.
12. Vorontsov I. E., Eliseeva I. A., Zinkevich A. Nikonov, M. Abramov, S. Boytsov, A. Kamenets, V. Kasianova, A. Kolmykov, S. Yevshin, et al. HOCOMOCO in 2024: a rebuild of the curated collection of binding models for human and mouse transcription factors. Nucleic Acids Res. 2024;52(D1):D154–D163.